
AI as a Medical Device: The Global Map of Regulation, Registration, and Market Access
Artificial intelligence is the fastest-growing class of medical device in history — and the most regulation-divergent. The U.S. FDA now lists 1,524 AI-enabled devices; South Korea authorized 153 in a single year. Yet the same software is Class II in the U.S., Class IIa+ and 'high-risk' in the EU, and Class III in China — each with its own evidence, local-holder, and change-control rules. This report maps how every major regulator classifies, approves, and polices AI as a medical device, with registration costs, timelines, and reliance routes across 30+ markets, and the playbook for reaching them without rebuilding the dossier each time.
An evidence-based field guide to how the world's regulators classify, approve, and police artificial intelligence in healthcare — and how to reach 30+ markets without rebuilding the dossier each time.
TL;DR
Artificial intelligence has become the fastest-growing category of medical device in history. The U.S. Food and Drug Administration's public list of AI-enabled devices reached 1,524 authorizations by mid-2026, with roughly three-quarters in radiology (FDA). On our own analysis of the FDA 510(k) database, AI's share of all clearances climbed from about 1 in 700 in 2019 to roughly 1 in 28 in 2025. South Korea alone authorized 153 AI devices in 2025 (MFDS).
But the same software that sails through one agency can stall in the next. Here is the argument of this report, in five points:
- AI is a device, and most of it is "Software as a Medical Device" (SaMD). When software diagnoses, triages, or recommends treatment, it is regulated like a scalpel or a scanner — in dozens of jurisdictions, each with its own rules.
- The whole world up-classifies it. The EU's MDR Rule 11, Brazil's Rule 11, China's Class III default for diagnostic AI — diagnostic and therapeutic software almost everywhere lands in a higher-risk class that demands third-party review.
- Adaptive AI broke the one-time-approval model. A model that keeps learning changes after it ships, so regulators invented new mechanisms — the U.S. Predetermined Change Control Plan, Japan's IDATEN, Korea's DMPA — that do not match one another.
- Reliance converges, AI diverges. A web of reliance and recognition pathways is supposed to let one approval unlock many markets. It works — for static devices. For adaptive AI, "a PCCP authorized by the FDA does not satisfy EU AI Act obligations, and vice versa" (Berkley Lifesciences).
- So the winners industrialize multi-market registration. The competitive edge is no longer a single clearance; it is the operational machine that turns one approval into thirty, and keeps each one valid as the algorithm evolves.
This is the gap Pure Global closes — in-country representation and AI-assisted regulatory execution across 30+ markets, on a flat annual fee. The rest of this report is the map.
What "AI as a Medical Device" actually is
Start with the word that does the heavy lifting: device. If a piece of software is "intended for use in the diagnosis, treatment, or prevention of disease," it is a medical device in almost every legal system on earth — whether it runs on a chip inside an MRI scanner or as an app in a radiologist's browser. AI does not get its own statute in most countries; it inherits the entire apparatus of medical-device law.
The anchor definition comes from the International Medical Device Regulators Forum (IMDRF), the body where the major agencies harmonize their vocabulary. In its foundational 2013 document, IMDRF defined Software as a Medical Device (SaMD) as "software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device" (IMDRF N10). The FDA adopted that language verbatim and draws a three-way distinction that matters for everything downstream (FDA):
- SaMD — software that is the medical device (a chest-X-ray triage algorithm, a diabetic-retinopathy detector). This is where most clinical AI lives.
- SiMD — software in a medical device, integral to hardware (the firmware running an infusion pump).
- Software used to manufacture or maintain a device, which is regulated differently again.
For AI specifically, IMDRF's 2022 key-terms document defines a Machine Learning-enabled Medical Device as "a medical device that uses machine learning, in part or in whole, to achieve its intended medical purpose" (IMDRF N67).
The distinction that breaks everything: locked vs. adaptive
Traditional device regulation rests on a simple bargain: you prove a device is safe and effective once, at a fixed design, and that design then stays put. AI shatters that assumption. The FDA's pivotal 2019 discussion paper drew the line precisely. A "locked" algorithm is one that "provides the same result each time the same input is applied to it and does not change with use" — a look-up table, a decision tree, a frozen classifier (FDA, 2019). An adaptive or continuously-learning algorithm changes after it is deployed.
That single property — software that improves itself in the field — is the reason AI needed a decade of new regulation. If the product you approved in January is not the product running in a hospital in June, what exactly did you approve? Every framework in this report is, at bottom, an attempt to answer that question, and the Total Product Life Cycle (TPLC) — oversight across the whole life of the device, not just the moment of clearance — is the shared response.
How regulators decide how hard to look
IMDRF's 2014 risk-categorization framework set the logic the world now follows: the scrutiny a SaMD deserves scales with two factors — the significance of the information it provides (does it inform, drive, or diagnose/treat?) and the severity of the healthcare situation (non-serious, serious, or critical) (IMDRF N12). An app that suggests stretches for back pain and an algorithm that flags a brain bleed are not the same regulatory animal, and this two-axis grid is why.
The World Health Organization added the ethical scaffolding. Its 2021 report Ethics and governance of artificial intelligence for health set six principles — protect autonomy; promote well-being and safety; ensure transparency and explainability; foster responsibility and accountability; ensure inclusiveness and equity; and promote responsive, sustainable AI (WHO). WHO followed with regulatory considerations in 2023 and, in January 2024, the first global guidance aimed squarely at generative AI and large multi-modal models (WHO).
How we got here
AI medical devices did not arrive with a single breakthrough; they accreted, country by country, over roughly a decade. The milestones below trace how a definition in 2013 became dedicated lifecycle rules on five continents.
A global timeline of AI medical-device regulation (2013–2026)
In barely a decade, AI SaMD went from an IMDRF definition to dedicated lifecycle rules on five continents.
| Date | Milestone |
|---|---|
| Dec 2013 | IMDRF N10 defines "Software as a Medical Device" (SaMD) |
| Jan 2017 | FDA clears Arterys — first cloud + deep-learning clinical tool (510(k)) |
| Apr 2018 | FDA authorizes IDx-DR — first autonomous AI diagnostic (De Novo) |
| May 2018 | Korea MFDS approves VUNO Med-BoneAge — Korea's first AI device |
| Dec 2018 | Japan PMDA approves EndoBRAIN (Class III) — Japan's first AI SaMD |
| Apr 2019 | FDA discussion paper on modifications to AI/ML SaMD (locked vs adaptive) |
| 2020 | China NMPA approves DeepVessel FFR — first Class III AI device; Japan launches IDATEN + DASH for SaMD |
| Jan 2021 | FDA AI/ML Action Plan; Jun 2021 WHO six ethics principles |
| Oct 2021 | GMLP — 10 guiding principles (FDA + Health Canada + UK MHRA) |
| 2022 | Saudi SFDA MDS-G010 — early dedicated AI device guidance (cited by some as the first "enforceable"); IMDRF N67 ML terms; Brazil RDC 751/657 |
| Aug 2024 | EU AI Act enters into force; WHO LMM (generative-AI) guidance (Jan 2024) |
| Dec 2024 | FDA finalizes Predetermined Change Control Plan (PCCP) guidance |
| Jan 2025 | IMDRF N88 GMLP final; Korea issues world's first generative-AI device guideline; FDA AI lifecycle draft guidance |
| Feb 2026 | IMDRF N89 Reliance Playbook |
| Apr 2026 | Health Canada finalizes ML-device guidance; Korea approves first generative-AI device |
Source: Compiled from IMDRF, US FDA, EU, NMPA, PMDA, MFDS, SFDA, ANVISA, Health Canada and WHO primary documents — Pure Global, June 2026.
A few moments deserve emphasis. In January 2017, the FDA cleared Arterys — the first clinical tool to combine cloud computing and deep learning (PR Newswire). Then, in April 2018, came the watershed: the FDA authorized IDx-DR, the first AI permitted to deliver a diagnosis autonomously, with no physician interpreting the result — a diabetic-retinopathy screen for primary care that hit 87.2% sensitivity and 90.7% specificity in its pivotal trial (npj Digital Medicine). Within months, Korea (VUNO Med-BoneAge, May 2018) and Japan (EndoBRAIN, December 2018) approved their own firsts; China followed in 2020 with DeepVessel FFR, its first Class III AI device (npj Digital Medicine).
The regulatory scaffolding caught up between 2021 and 2025: the trilateral Good Machine Learning Practice principles in October 2021, the EU's AI Act entering into force in August 2024, the FDA's final Predetermined Change Control Plan guidance in December 2024, and — telling you how fast the frontier moves — Korea's world-first generative-AI device guideline in January 2025 (BioWorld). By February 2026, IMDRF had published a global Reliance Playbook to help regulators lean on one another's work (IMDRF N89).
The data: how big, how fast, where
The clearance curve is bending sharply upward
The single best gauge of AI's penetration into medicine is the FDA's public Artificial Intelligence-Enabled Medical Devices list, which reached 1,524 authorizations on its most recent update (reflecting data through the first quarter of 2026) (FDA). To see the velocity, we analyzed the underlying FDA 510(k) database directly. Identified AI/ML clearances rose from a handful in the late 2010s to 115 in 2025 — and, more tellingly, AI grew from 0.14% of all 510(k) clearances in 2019 to 3.57% in 2025, a roughly 25-fold jump in share in six years.
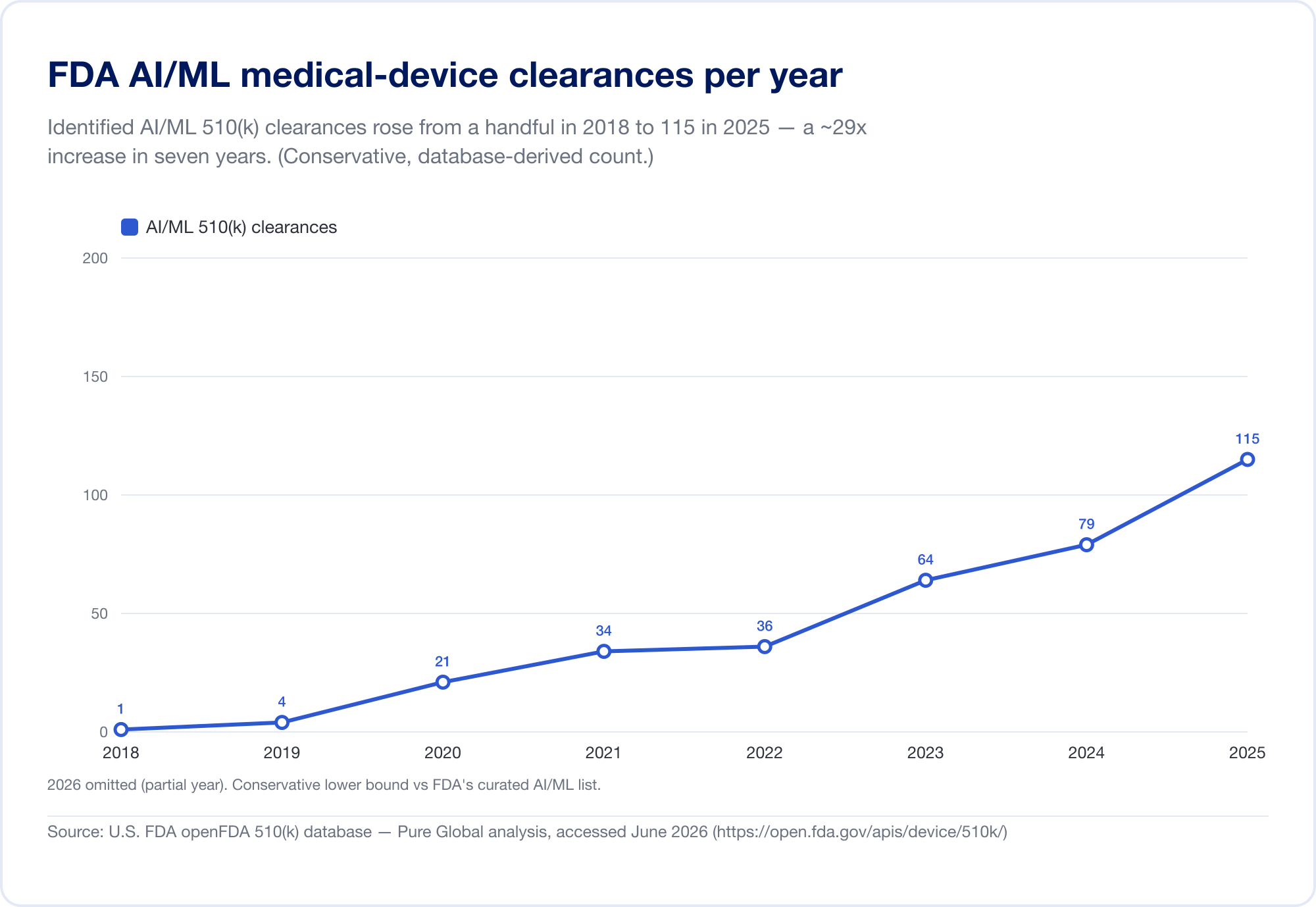
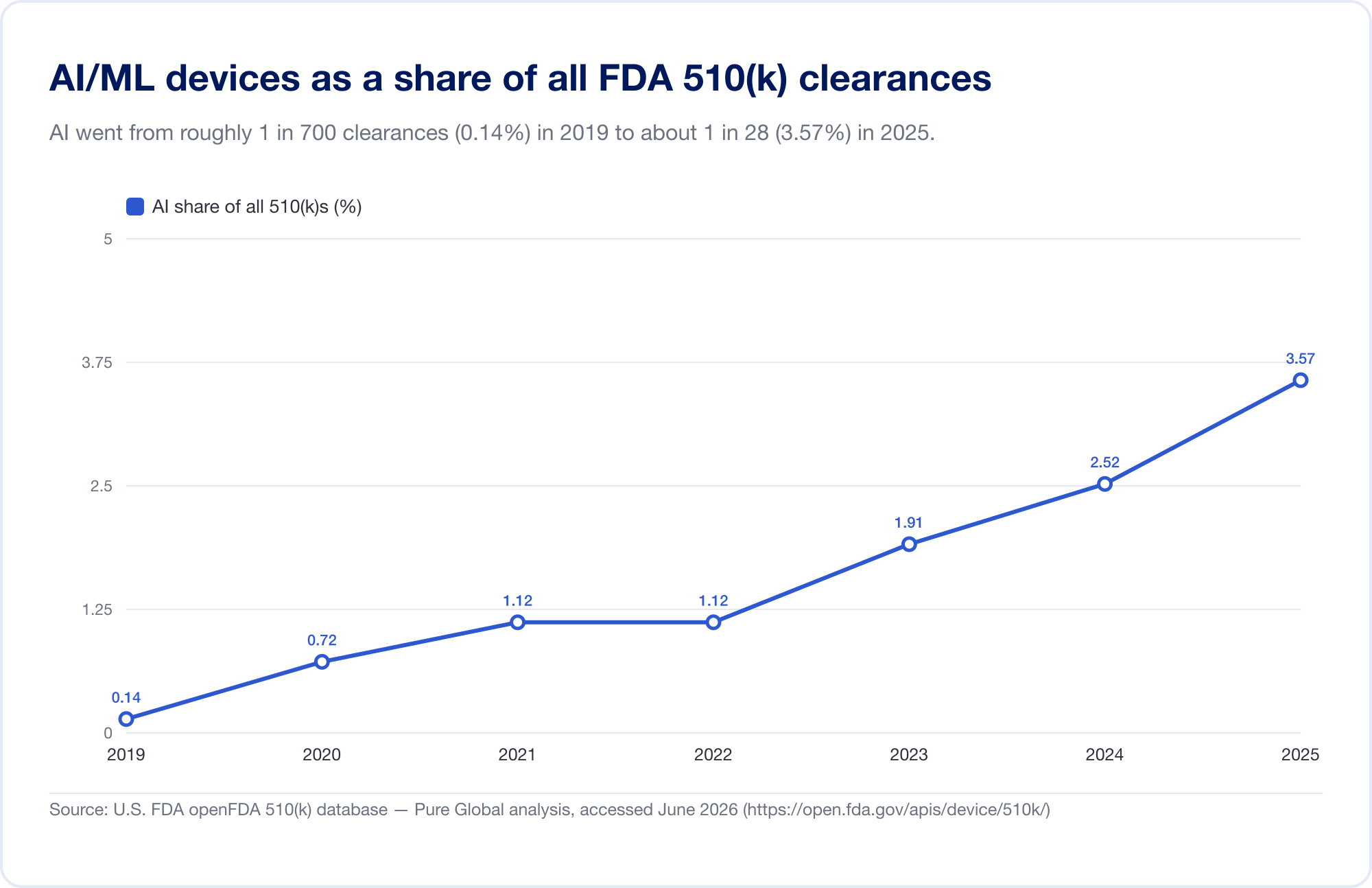
(Our database-derived count is deliberately conservative — narrower than the FDA's curated list, because many AI radiology tools sit under product codes whose text never says "AI." We use it for trend and geography; the FDA's own list is the headline total. Source: openFDA 510(k) database — Pure Global analysis, June 2026.)
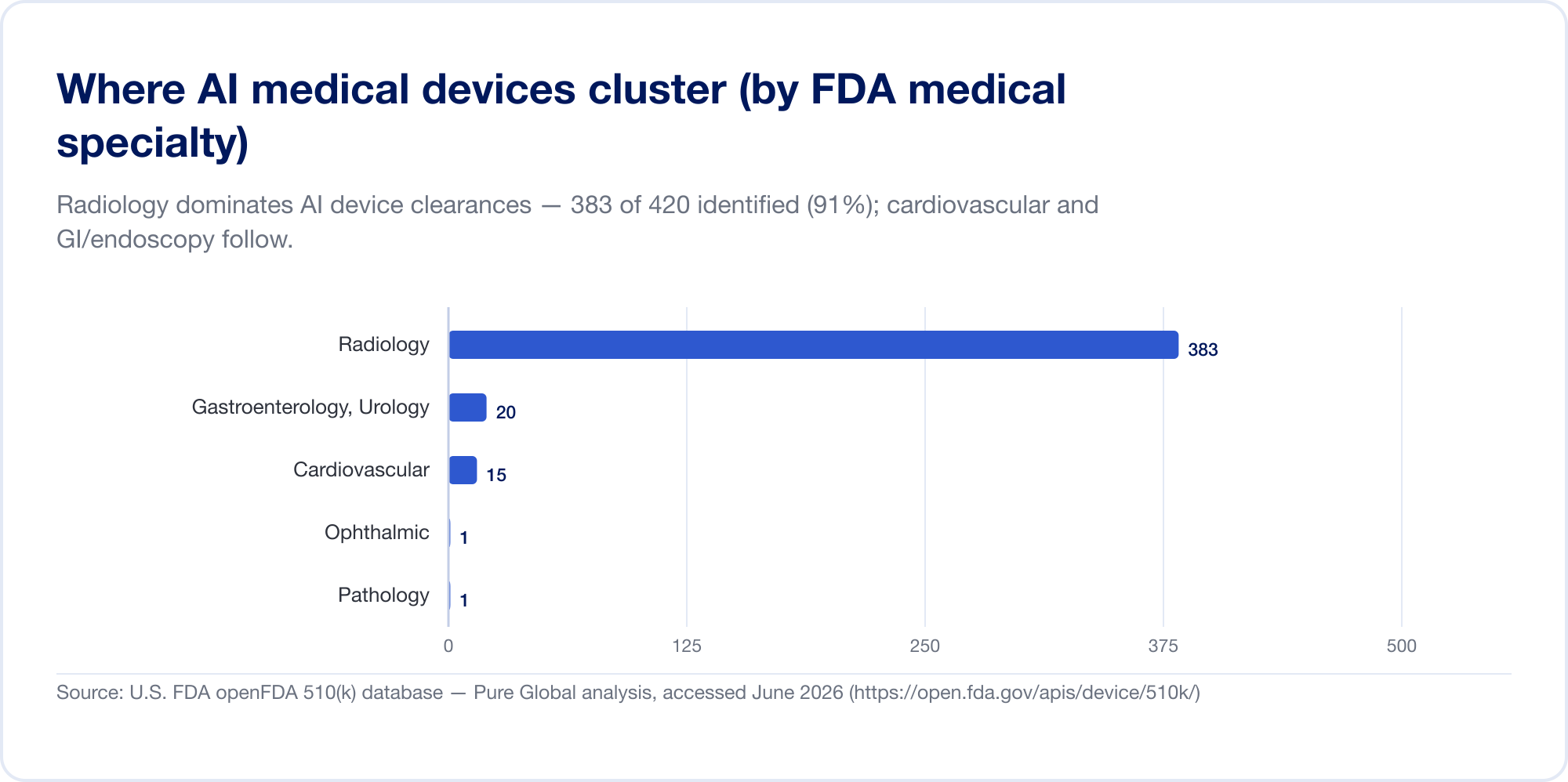
It is overwhelmingly an imaging story — for now
AI in medicine is, today, mostly AI in radiology. On the FDA's list, radiology accounts for about 76% of all AI authorizations (The Imaging Wire); in our own clearance sample the concentration is even higher, with cardiovascular and gastroenterology (endoscopy) a distant second and third. The reason is structural: imaging is digital, abundant, and labelled, and the 510(k) pathway lets a new algorithm cite an existing one as a predicate. Pathology, cardiology signals, and clinical-text models are growing, but the centre of gravity remains the image.
The innovators are everywhere; the markets are everywhere else
Here is the fact that should reframe any commercialization plan. AI medical devices cleared in the U.S. come from at least 16 countries. In our analysis of AI 510(k) applicants, the United States leads, but South Korea, Israel, Canada, France, China, Germany, the United Kingdom, the Netherlands, Taiwan, and Japan each field meaningful numbers.
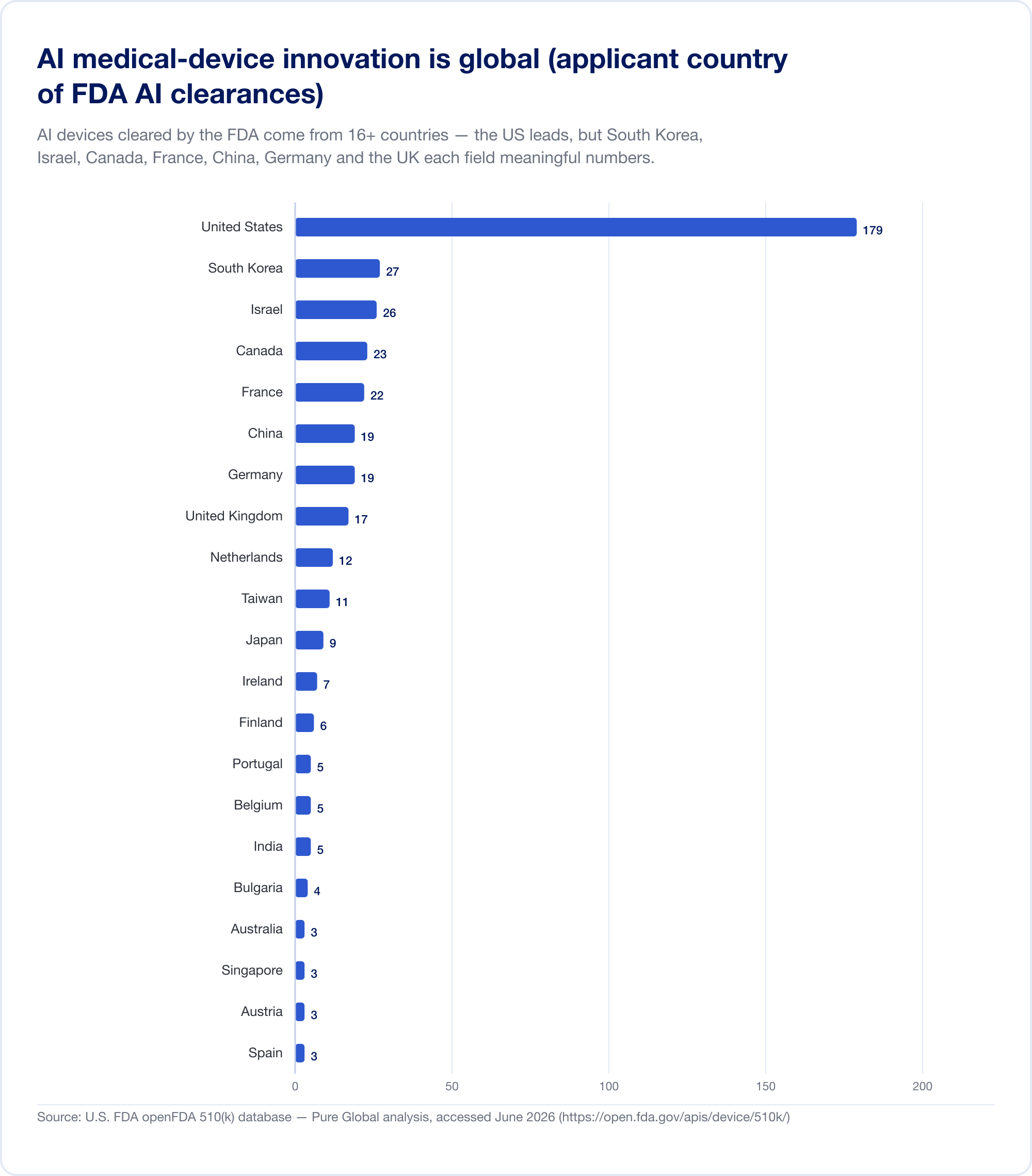
National output confirms the pattern. South Korea authorized (approvals and certifications combined) 64 AI devices in 2023, 108 in 2024, and 153 in 2025 — a 41.6% jump, with 77.7% domestically made (MFDS). China had approved roughly 154 AI medical devices by mid-2025, about 80% of them in the highest-risk Class III (JMIR Medical Informatics). Taiwan licensed 166 AI/ML devices between 2020 and 2024 (J. Formos. Med. Assoc.). Japan had 51 AI-based SaMD on the PMDA's list as of September 2025 (Global Health & Medicine).
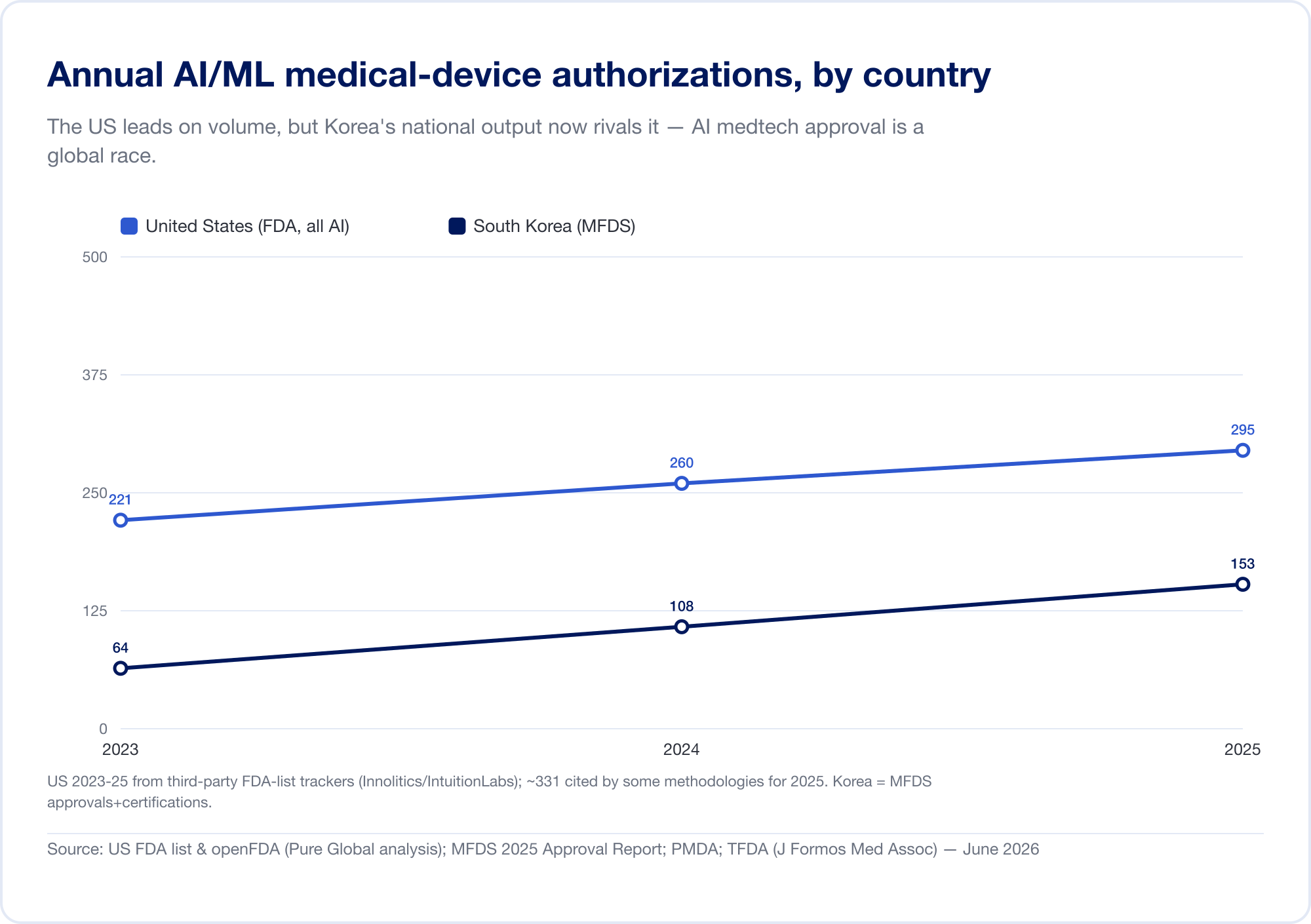
The implication is direct: a brilliant algorithm built in Tel Aviv, Seoul, or Shanghai has to reach patients in markets that each speak a different regulatory language. Innovation is global; approval is stubbornly local.
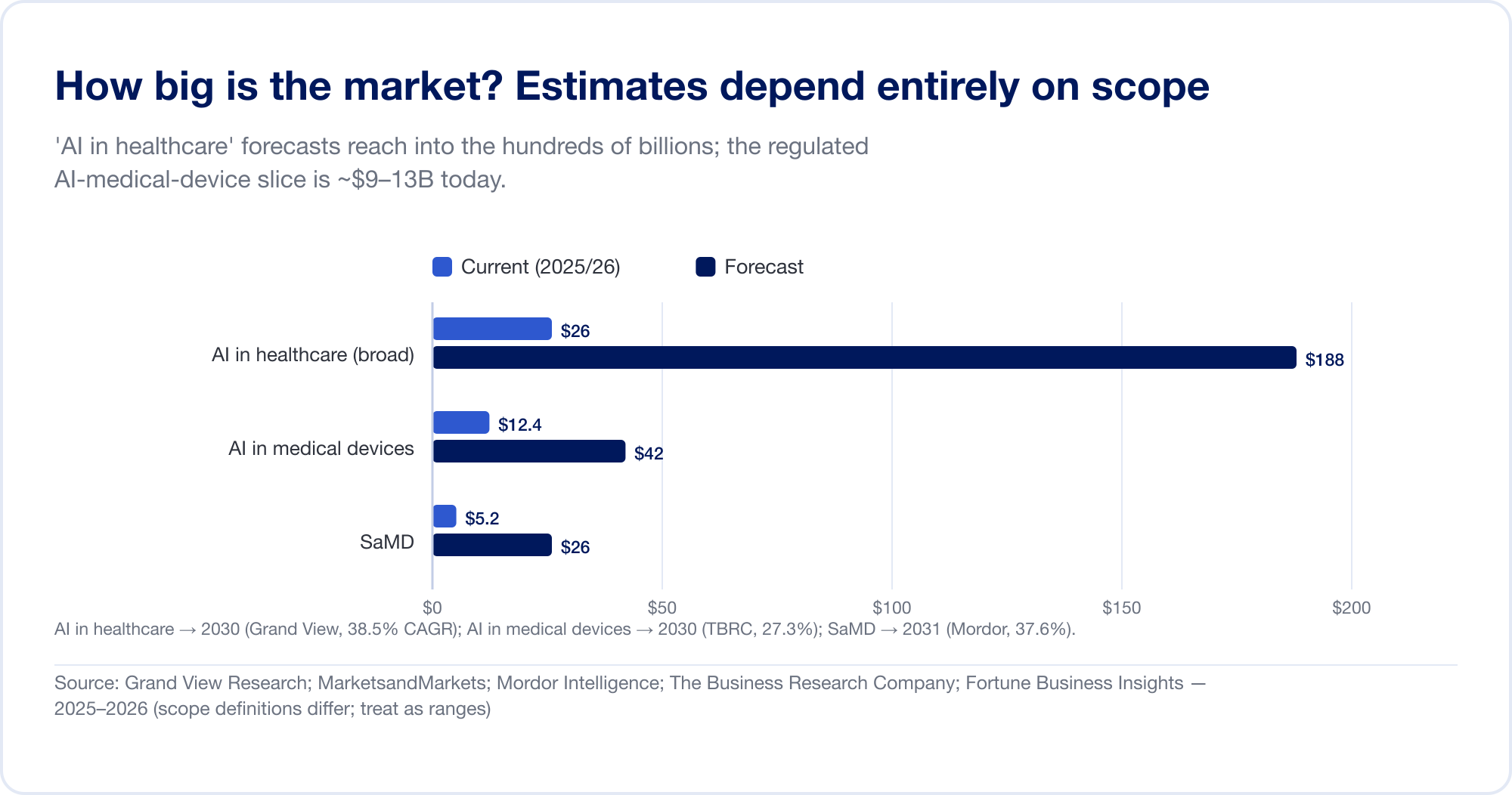
How big is the market? It depends entirely on what you count
Market-size estimates for AI in healthcare span an order of magnitude, because analysts draw the boundary in wildly different places. The broad "AI in healthcare" category — which bundles drug discovery, administrative automation, ambient scribes, and more — is forecast at USD 187.7 billion by 2030 at a 38.5% CAGR (Grand View Research), with the most aggressive house projecting over USD 1 trillion by 2034 (Fortune Business Insights). The narrower, regulated slice — AI in medical devices — is far smaller and more credible: about USD 12.4 billion in 2025, reaching USD 42.4 billion by 2030 (The Business Research Company). Pure SaMD is estimated at roughly USD 5–25 billion depending on the source (Mordor Intelligence). Treat every one of these as a range, not a fact.
The capital tells a cleaner story. U.S. digital-health venture funding rebounded to USD 14.2 billion in 2025 (+35%), and for the first time AI-enabled startups captured the majority — 54% of all dollars (Rock Health). Public AI-SaMD names are inflecting from growth to profitability: Tempus AI posted USD 1.3 billion in FY2025 revenue (+83%) (Tempus); iRhythm reached USD 747 million and its first GAAP-profitable quarter (iRhythm); HeartFlow went public in August 2025 and grew 40% to USD 176 million (HeartFlow); Korea's Lunit grew 53% with 92% of revenue from overseas (Lunit). Among private players, Aidoc raised a USD 150 million Series E in April 2026, taking total funding past half a billion dollars (Aidoc).
The patent race has a different leader
Intellectual property is where the global contest is most visible. WIPO's landmark study counted roughly 340,000 AI-related patent filings, with life and medical sciences the third-largest application field (WIPO). But the geography has since flipped: in the decade to 2023, China filed 38,210 generative-AI inventions — more than the rest of the world combined — against 6,276 from the United States (WIPO). China's patent office issued dedicated AI patent-examination guidelines on the last day of 2024 (CNIPA). The pipeline of medical AI is being filled disproportionately from Asia — which makes multi-market access strategy a question of when, not if, for a growing share of the world's developers.
The blind spot: you cannot see SaMD in trade data
One analytical caution that reframes how to think about this market. Traditional medtech market intelligence leans on import and customs data — but pure SaMD is invisible to it. Software delivered by cloud download or app store crosses no physical border, generates no HS-code customs declaration, and is not captured in merchandise-trade statistics. The international standard-setters say so directly: "Data flows that are not directly monetised are not generally considered as trade flows in current statistical standards" (OECD-IMF-WTO), and the WTO's moratorium on customs duties for electronic transmissions has held since 1998 (UNCTAD). AI-embedded hardware — an AI-enabled CT scanner — moves as a good and shows up in the data; a 510(k)-cleared cloud algorithm does not. The upshot: conventional trade analysis systematically under-counts AI software and over-weights hardware. The only reliable footprint of a SaMD's global spread is its registrations — which is exactly the lens this report uses.
The safety reckoning
Behind every tightening of the rules is a body of evidence that AI in medicine can fail in ways traditional devices do not. Three findings, in particular, reshaped how regulators think.
Bias that hides in plain data. A landmark New England Journal of Medicine study found that pulse oximeters — ubiquitous, and increasingly paired with algorithms — missed dangerously low blood-oxygen levels (occult hypoxemia) in 11.7% of Black patients versus 3.6% of White patients, a roughly threefold disparity across some 50,000 paired readings (NEJM, 2020). The FDA issued a safety communication in February 2021 and, by January 2025, draft guidance demanding more diverse validation across skin tones (FDA). The lesson generalized: a model trained on a non-representative population can carry that bias silently into every hospital that deploys it.
Validation that doesn't survive contact with reality. The Epic Sepsis Model — a proprietary prediction tool deployed at hundreds of U.S. hospitals — was externally validated on 38,455 hospitalizations and scored an area under the curve of 0.63, far below the vendor's claimed 0.76–0.83. It missed 67% of sepsis cases while firing alerts on 18% of all patients (JAMA Internal Medicine, 2021). A model can be "in production" at scale and still not work as advertised.
Recalls that cluster right after clearance. A 2025 Johns Hopkins/Yale analysis of FDA-authorized AI devices found that 43.4% of AI-device recalls occurred within the first 12 months of clearance — roughly double the rate for 510(k) devices overall (JAMA Health Forum, 2025). A parallel study found recalls concentrated among devices that lacked published clinical studies (JAMA Network Open, 2025). Context matters: roughly 97% of AI/ML devices clear via the 510(k) pathway, which requires no prospective human testing — so a great deal rides on post-market vigilance.
The safety reckoning: why regulators are tightening on AI
Bias, validation gaps and early recalls are the evidence behind the shift to lifecycle oversight.
| Finding | Figure | Source |
|---|---|---|
| Pulse-oximeter occult hypoxemia, Black vs White patients | 11.7% vs 3.6% (~3x) | NEJM, Dec 2020 |
| Epic Sepsis Model external AUC (vs 0.76–0.83 claimed) | 0.63; missed 67% of sepsis cases | JAMA Intern. Med., Jun 2021 |
| AI-device recalls within 12 months of clearance | 43.4% (~2x all 510(k)s) | JAMA Health Forum, 2025 |
| AI devices recalled (903 studied) | 4.8%, concentrated in those lacking clinical studies | JAMA Network Open, 2025 |
| AI devices cleared via 510(k) (no prospective testing required) | ~97% | Analyses of FDA list, 2025 |
Add to this the problem of dataset shift — deployed models silently degrade as the patient population, scanners, or coding systems around them change — and you have the rationale for the entire modern apparatus: Good Machine Learning Practice, Predetermined Change Control Plans, and mandatory real-world performance monitoring. Regulators are not tightening because AI doesn't work. They are tightening because it works until it quietly doesn't.
The global regulatory map
This is the reference core of the report: how the major jurisdictions actually classify, review, and police AI SaMD as of mid-2026. The throughline to watch is classification (which risk class the software lands in) and change control (what happens when the algorithm updates). A consolidated comparison matrix follows the regional detail.
How 15 jurisdictions regulate AI as a medical device (2026)
Same software, fifteen answers: classification, AI-specific guidance, and change-control rules diverge market by market.
| Jurisdiction | Where most AI SaMD lands | Dedicated AI/SaMD guidance | Change-control / adaptive-AI mechanism | Reliance on foreign approval |
|---|---|---|---|---|
| United States (FDA) | Class II (510(k)/De Novo) | Yes — PCCP final 2024; lifecycle draft 2025 | PCCP (pre-authorize changes, no new submission) | No (own review; predicate-based) |
| European Union | Class IIa+ (MDR Rule 11) + AI Act high-risk | MDR + AI Act + MDCG 2025-6 | Substantial change → Notified Body re-review + AI Act | No (CE conformity assessment) |
| United Kingdom (MHRA) | Class IIa+ (UK MDR 2002) | Software & AI Change Programme; AI Airlock | PCCP planned (statutory instrument) | CE accepted in GB to 2028/2030 |
| Canada (Health Canada) | Class II–IV | Yes — ML guidance final Apr 2026 | PCCP | MDSAP for QMS; not full product reliance |
| Australia (TGA) | Class IIa–III | AI review 2024 (14 findings) | Under development | Comparable-overseas-regulator route |
| Japan (PMDA/MHLW) | Class II–III | DASH for SaMD; SaMD guidance | IDATEN (PACMP) pre-agreed changes | Foreign clinical data accepted; MDSAP |
| South Korea (MFDS) | Grade 2–3 | Yes — incl. world-first generative-AI guideline | DMPA pre-approved change plans | Limited; own review |
| Singapore (HSA) | Class B–D | Yes — GL-04-R4 (2025), AI-MD lifecycle | Change Notification | Yes — 5 reference agencies (~98% abridged) |
| China (NMPA) | Class III (decision software) | Yes — CMDE AI principles + classification catalog | Carve-out only if core algorithm unchanged | No (in-country agent; type testing) |
| India (CDSCO) | Class A–D | Draft only (Oct 2025) | Algorithm Change Protocol (proposed) | Reference-country approval eases Class C/D |
| Taiwan (TFDA) | Class II–III | Yes — CADe/CADx + PCCP-drafting guidance | PCCP guidance (2024) | Local performance evaluation emphasized |
| Brazil (ANVISA) | Class II–IV (Rule 11) | RDC 657 (SaMD); no dedicated AI rule | Full change registration | Yes — IN 290/2024 (Class III/IV, 4 agencies) |
| Mexico (COFEPRIS) | Class I–III | General device rules | Re-registration | Yes — abbreviated pathway (IMDRF + MDSAP) |
| Saudi Arabia (SFDA) | Class A–D | Yes — MDS-G010 (early; cited as first "enforceable") | Change notification via GHAD | Supportive only; local validation required |
| UAE (EDE) | Class I–III | General device rules | Re-registration | Yes — recognizes CE/FDA |
Source: US FDA, EU MDR/AI Act, MHRA, Health Canada, TGA, PMDA/MHLW, MFDS, HSA, NMPA, CDSCO, TFDA, ANVISA, COFEPRIS, SFDA, EDE — Pure Global analysis, June 2026.
United States — the benchmark, and the busiest
The U.S. runs the world's most active AI device regime, administered by the FDA's Center for Devices and Radiological Health and its Digital Health Center of Excellence. There is no special "AI statute"; AI functions that meet the device definition are regulated as SaMD through three pathways: 510(k) clearance (demonstrating "substantial equivalence" to a predicate device), De Novo classification (for novel low-to-moderate-risk devices with no predicate), and PMA (premarket approval, for the highest-risk Class III). The overwhelming majority of AI devices — about 97% — enter via 510(k); only a couple of dozen have used De Novo, and a handful PMA (analysis of FDA list). The landmark De Novo was IDx-DR in 2018.
The defining recent development is the Predetermined Change Control Plan (PCCP), finalized in December 2024 (FDA). A PCCP lets a manufacturer pre-specify and pre-authorize a set of future model modifications — and the agency's own words capture the value: the FDA reviews the PCCP "to ensure the continued safety and effectiveness of the device without necessitating additional marketing submissions for implementing each modification." In January 2025 the FDA went further, issuing comprehensive draft guidance on the total product life cycle of AI-enabled device software. The U.S. posture, in short: fast, predicate-driven, imaging-heavy, and now organized around lifecycle oversight.
European Union — two regimes stacked on one device
The EU is the hardest major market for AI SaMD, because two regulatory regimes now apply at once. First, the Medical Device Regulation (MDR). Its software classification rule — Rule 11 — is notorious. Read it directly: "Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa," escalating to Class III where a wrong decision could cause death or irreversible deterioration, or Class IIb for serious deterioration (MDR, Annex VIII, via EUR-Lex). Under the old directives most standalone software self-certified as Class I, with no third party involved. Rule 11 pushed nearly all diagnostic and therapeutic SaMD up to Class IIa or higher, which forces a Notified Body conformity assessment, ISO 13485 quality management, and clinical evaluation. Our own scan of the EUDAMED database found AI-keyword devices concentrated in Class IIa — exactly the Rule 11 footprint.
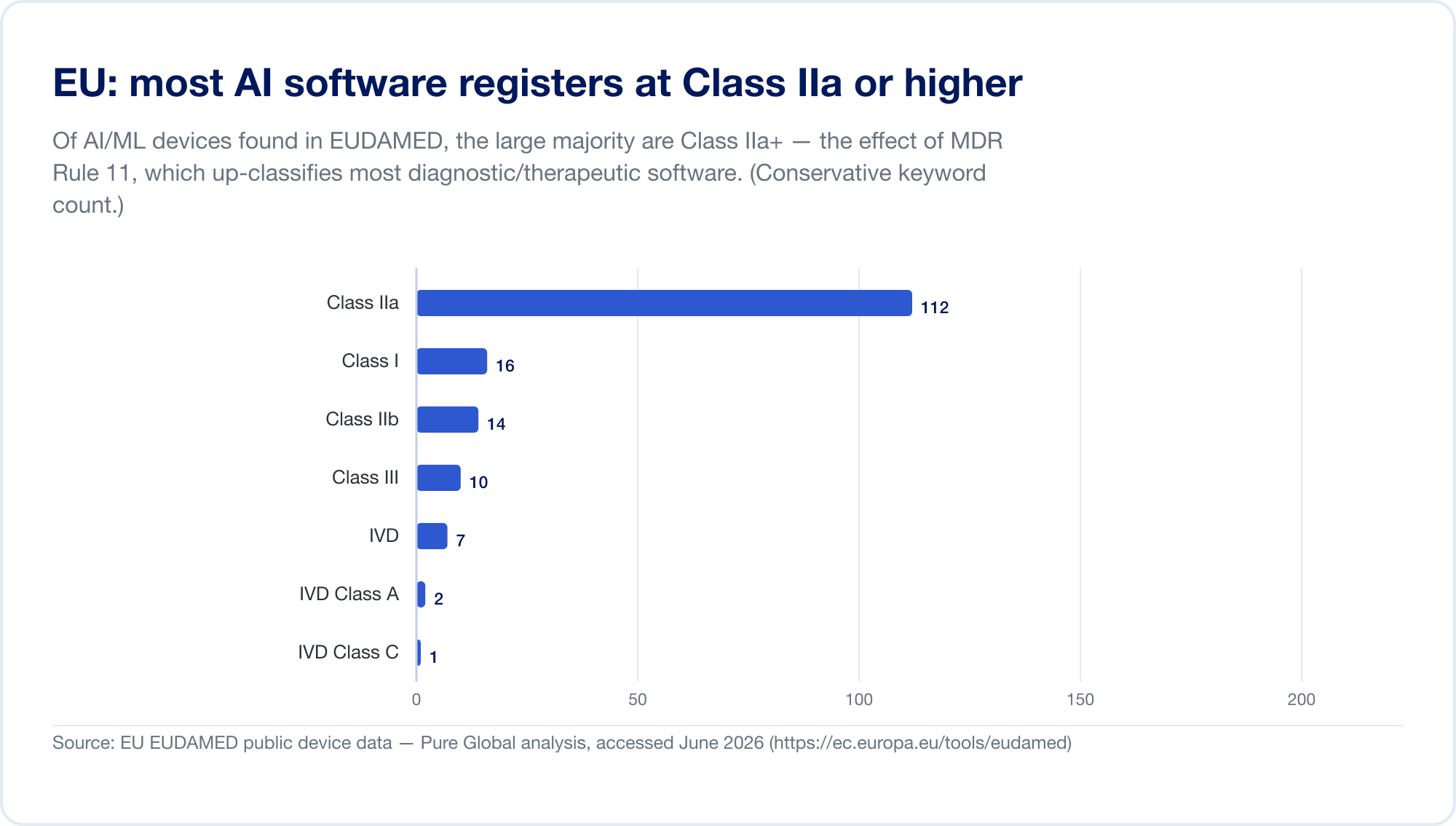
Then, layered on top, the EU AI Act (Regulation 2024/1689), in force since 1 August 2024. Through Article 6(1), an AI system is "high-risk" when it is (or is a safety component of) a product that already requires third-party conformity assessment — which captures essentially all Class IIa+ AI medical devices. High-risk obligations phase in through 2 August 2026, with the date for AI embedded in MDR/IVDR devices set at 2 August 2027 (a November 2025 "Digital Omnibus" proposal may defer this to 2028 — treat the dates as moving) (artificialintelligenceact.eu). Penalties run up to €35 million or 7% of global turnover (Article 99). To clarify the overlap, the MDCG and the new European AI Board issued a joint FAQ, MDCG 2025-6, in June 2025 (European Commission).
The binding constraint is capacity. The number of MDR Notified Bodies fell from roughly 80–96 under the directives to about 50, with only ~17–19 designated under the IVDR; MDR certification now takes 13–18 months on average, roughly double the pre-MDR norm (MedTech Europe). For AI SaMD, every device competes for scarce Notified Body slots — and after 2027/28 will need AI Act conformity assessment as well.
United Kingdom — diverging, pragmatically
Post-Brexit, the MHRA has charted a deliberately innovation-friendly course. Its Software and AI as a Medical Device Change Programme (roadmap published October 2022) spans eleven work packages, and the agency has committed to permitting PCCPs in forthcoming pre-market rules (MHRA). Its AI Airlock regulatory sandbox — the first of its kind for AI medical devices — ran a four-project pilot in 2024 and a seven-technology Phase 2 through 2026, with multi-year funding now committed (MHRA). Practically, about 90% of devices on the British market still carry the CE mark, which Great Britain will accept until 2028–2030; the MHRA consulted in early 2026 on recognizing CE marks indefinitely (MHRA).
Canada — first to finalize dedicated ML rules
Health Canada was among the first regulators with finalized dedicated pre-market guidance for machine-learning-enabled medical devices — first finalized in 2025 and published in revised final form in April 2026 — covering Class II–IV, adopting the IMDRF key terms, and formally introducing the PCCP so that authorized changes do not trigger a new licence amendment (Health Canada). Canada co-authored the foundational tri-regulator documents — GMLP (2021), PCCP principles (2023), and Transparency principles (2024) — and has required MDSAP certification since 2019.
Australia — reformed early, recalibrating for AI
Australia's TGA reformed its software rules back in February 2021, carving out low-risk wellness apps while up-classifying diagnostic software (active devices for therapy with a diagnostic function moved to Class III). Its 2024 consultation, Clarifying and strengthening the regulation of AI, drew over 600 stakeholders and produced 14 key findings now being worked through (TGA). The TGA's reliance route — accepting "comparable overseas regulators" — is a major accelerant, discussed below.
Japan — built for iteration
Japan regulates SaMD as "programmed medical devices" and has arguably the most AI-update-friendly mechanism of any major market: IDATEN, in force since September 2020, is Japan's version of a post-approval change-management protocol, letting manufacturers pre-agree changes to frequently-updated AI (PMDA). Combined with the DASH for SaMD initiative and the SAKIGAKE priority pathway, Japan has built infrastructure for software that evolves — though uptake is modest: just 51 AI-based SaMD on the PMDA's list as of September 2025.
South Korea — the fastest mover
Korea is the standout. Beyond its 153 AI authorizations in 2025, it has built a purpose-made legal framework: the Digital Medical Products Act (DMPA), effective January 2025, introduces a PCCP-style change mechanism and a Digital QMS aligned to IMDRF work items (Emergo). Korea also issued the world's first guideline for generative-AI medical devices in January 2025 and approved its first such device in April 2026 — and it chairs the IMDRF AI/ML working group. If you want to see where global AI regulation is heading, watch Seoul.
Singapore — the reliance hub
Singapore's HSA punches far above its size by being the most efficient reliance regime in Asia. Its software guidance, GL-04 (Revision 4, December 2025), explicitly covers machine-learning-enabled devices across the lifecycle and requires a Change Notification when an AI model's performance, inputs, or human-oversight level change (HSA). Crucially, HSA recognizes five reference agencies (US FDA, EU Notified Bodies, Health Canada, TGA, Japan MHLW) and has estimated that about 98% of applications can use an abridged route; devices with two prior approvals can register through an "immediate" route in as little as an hour (Singapore MOH).
China — large, distinct, and demanding
China's NMPA treats AI decision-support software seriously: its 2021 CMDE review principles and 2021–2022 classification catalog place software that delivers a diagnosis or drives treatment in Class III, the highest risk tier. China approved its first Class III AI device in 2020 and had reached roughly 154 AI medical devices by mid-2025 (JMIR Medical Informatics). In an October 2025 reform package, the NMPA committed to "simplify the change registration requirements for AI-powered medical devices where the core algorithm remains unchanged but algorithm performance is optimized" — a real but narrow concession compared with the U.S. PCCP (NMPA). China requires an in-country agent, local type-testing, and — for many Class III devices — local clinical data, making it among the most demanding markets in this report.
India, Taiwan, and the rest of Asia-Pacific
India's CDSCO issued a draft Medical Device Software guidance in October 2025, introducing an "Algorithm Change Protocol" for AI updates, but it remains a draft with no finalized AI rule yet (CDSCO). Taiwan's TFDA, by contrast, has one of the deepest AI guidance suites anywhere — dedicated CADe/CADx technical guidelines and PCCP-drafting guidance — and licensed 166 AI/ML devices from 2020 to 2024. Across ASEAN, the pattern is reliance-plus-localization: Malaysia, Thailand, Vietnam, the Philippines, and Indonesia mostly treat SaMD as a general device and lean on reference-country approvals, with Vietnam's fast-track unusually broad (it accepts even NMPA and MFDS approvals).
Latin America, the Middle East, and Africa
Brazil's ANVISA imported the EU's logic wholesale: its Rule 11 under RDC 751/2022 mirrors the EU's, placing decision-support software in Class II–IV, and RDC 657/2022 was the region's first SaMD-specific resolution. A foreign manufacturer cannot hold a Brazilian registration — a local Brazil Registration Holder is mandatory and non-waivable (Artixio). Mexico's COFEPRIS overhauled its reliance regime in 2025 into a single Abbreviated Pathway recognizing all IMDRF and MDSAP members, targeting 30 working days. Saudi Arabia's SFDA issued MDS-G010 (November 2022) — among the first dedicated AI/ML medical-device guidances anywhere, and cited by some observers as the first enforceable one (others classify it as non-binding) — which uniquely directs that "the manufacturer should locally validate the AI/ML-based medical devices that developed and approved in other jurisdictions" (SFDA) — a reminder that "reference-country approval" is not always enough. The UAE centralized device approval under a new Emirates Drug Establishment in January 2025. South Africa's SAHPRA issued its first AI communication in September 2025 but has not yet begun registering devices, and the continental African Medicines Agency — 31 of 55 states ratified — does not yet cover devices or AI.
This is the heart of the problem: fifteen jurisdictions, fifteen answers. The same software is Class II in the U.S., Class IIa+ and "high-risk" in the EU, Class III in China, Grade 2–3 in Korea, and Class II–IV in Brazil — each with its own evidence, language, local-holder, and change-control requirements.
What it costs and how long it takes
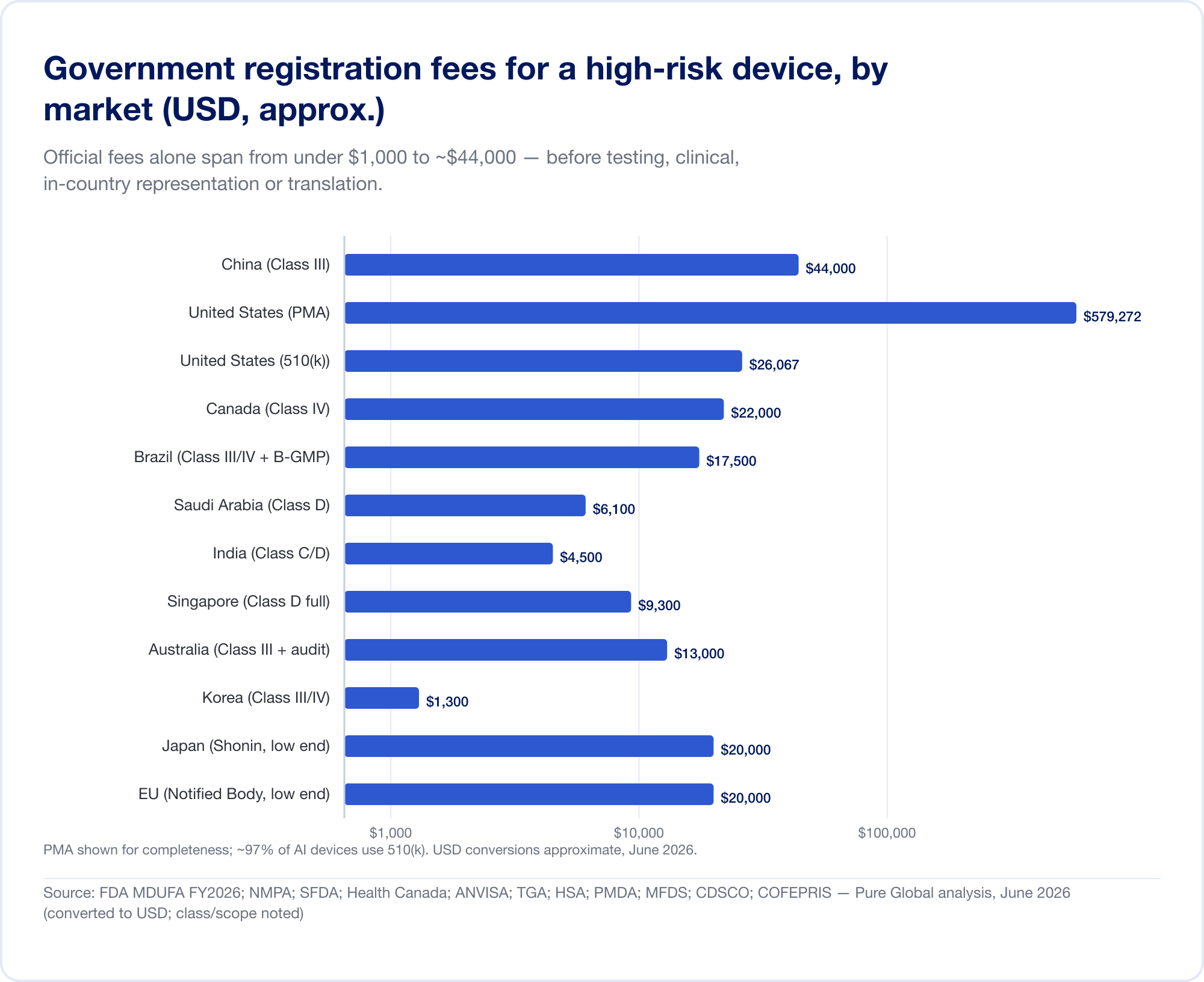
The classification differences above translate directly into money and months. The headline numbers below are government fees and realistic timelines for a higher-risk device; they exclude the substantial costs of testing, clinical evidence, translation, and in-country representation that often dwarf the official fee.
The official fee is the small number
Government fees alone range from under USD 1,000 to about USD 44,000 for a high-risk registration:
- United States — MDUFA FY2026 fees (verified on FDA.gov): 510(k) $26,067 (small-business $6,517); De Novo $173,782; PMA $579,272; plus an annual establishment fee of $11,423 (FDA).
- China — NMPA registration fees of roughly RMB 210,900 (~$30,000) for Class II and RMB 308,800 (~$44,000) for Class III — the steepest official fees in this report.
- Brazil — ANVISA Class III/IV registration of ~BRL 21,000 plus a B-GMP certification fee of BRL 72,804 for international manufacturers.
- Canada — Class III CAD $14,163, Class IV CAD $30,713 (April 2026).
- Saudi Arabia — SFDA fees of SAR 15,000–23,000 by class.
- India — MD-15 import licence of $3,000 per site + $1,500 per product for Class C/D.
- Singapore, Australia, Korea, Japan — official fees are comparatively modest (often under USD 13,000), but the evidence and review burden varies widely.
Time is the expensive number
The real cost is the calendar. Realistic, high-risk timelines stretch from about a month (Canada, Class III) to 24 months in the EU and four to five years in China when a local clinical trial is required:
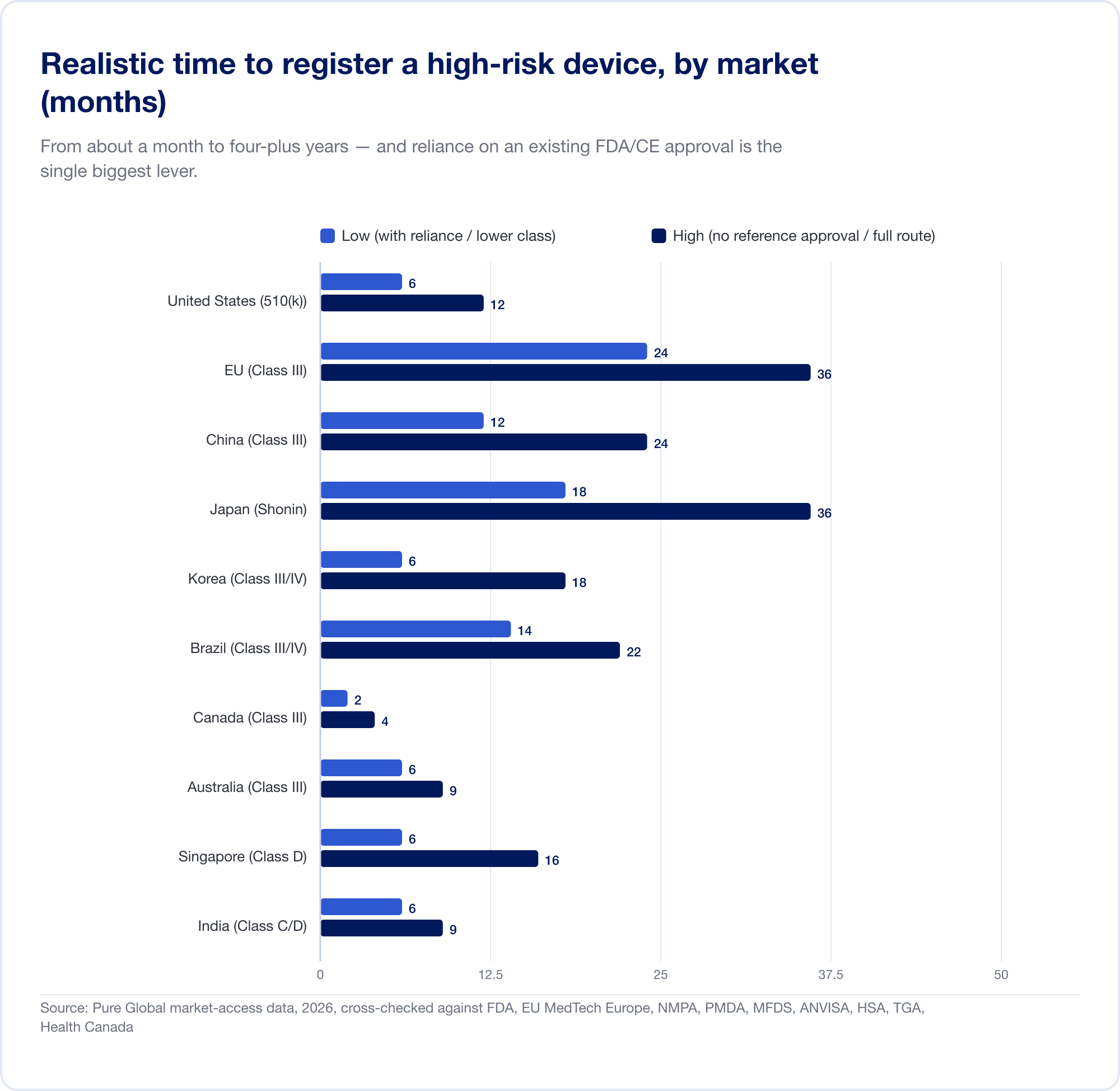
The EU illustrates how classification becomes cost. Because Rule 11 converts what used to be a near-free Class I self-declaration into a Class IIa+ Notified Body engagement, the all-in CE project for SaMD commonly runs into six figures and 13–18 months before a certificate issues. For a startup with a runway measured in quarters, that is not a line item — it is a strategic threat.
The lever: reliance, and the AI-specific twist
Against those timelines sits the single most powerful tool in global market access: reliance. Most markets outside the U.S., EU, and China will lean on an approval you already hold. Our own market data shows the effect starkly — a high-risk SaMD that takes ~8 months to register in Brazil on the standard route can drop toward 6 weeks when an FDA or other reference approval is leveraged through ANVISA's optimized-analysis pathway.
For AI SaMD there is a specific, recurring twist that the cost tables don't show. When the algorithm updates — as good AI constantly does — the question is whether that update needs a new submission. The U.S. PCCP can let pre-specified updates ship with no new submission, saving the $26,067 fee plus the 90–175-day review on each avoided 510(k), and far more for avoided PMA supplements. About 10% of the AI devices the FDA cleared in 2025 already embedded a PCCP. But that saving is U.S.-only — and that is the hinge of the entire report.
How much does it cost to register AI as a medical device in different countries?
The government fees above are only half the invoice. The other half is the specialist work — building the dossier, holding the local registration, fielding every question from the authority, and filing each renewal and algorithm change. This is where the industry is at its most opaque: most regulatory consultancies bill by the hour or quote each submission separately, so the true multi-market cost only emerges after the change orders arrive.
Pure Global is the first medical-device market-access firm to publish a single, flat annual fee per registration. From USD $2,000 per device, per market, per year, that one fee consolidates services normally billed hourly — in-country representation, submission (on a reference approval), renewals, modifications, and all health-authority correspondence. No timesheets, no per-email surprises.
Here is exactly what it costs to have Pure Global act as your local representative and hold an AI medical-device registration, market by market. (AI SaMD usually lands in the higher-risk class, so the upper figure applies in tiered markets.)
Pure Global in-country representation — flat annual fee per AI device
One transparent number per market, per year — everything for that registration included.
| Market | Local role Pure Global provides | Flat annual fee (USD) |
|---|---|---|
| United States | FDA US Agent | $1,000 |
| European Union | EU Authorized Representative | $2,000 |
| United Kingdom | UK Responsible Person (UKRP) | $2,000 |
| Australia | TGA Sponsor | $2,000 |
| Singapore | Registrant | $2,000 · $3,000 (Class C/D) |
| Malaysia | Authorized Representative | $2,000 · $3,000 (Class C/D) |
| Thailand | Authorized Representative | $2,000 · $3,000 (Class 3/4) |
| Indonesia | Authorized Representative | $2,000 |
| Vietnam | Market Authorization Holder | $2,000 |
| Hong Kong | Local Responsible Person | $2,000 · $3,000 (Class III/IV) |
| Macau | License holder & registration | $2,000 · $3,000 (Class III) |
| Brazil | Brazil Registration Holder (BRH) | $2,000 · $3,000 (Class III/IV) |
| Mexico | Mexico Registration Holder | $2,000 · $3,000 (Class II/III) |
| Colombia | INVIMA Representative | $2,000 · $3,000 (Class IIb/III) |
Flat annual fee for in-country representation of one AI SaMD; includes submission on a reference approval, renewals, modifications, and authority correspondence. Source: Pure Global Master Price List, 2026 (per registration; multi-registration and 3-year-contract discounts apply).
One-time submission and compilation work — where a market needs a full dossier built — is published just as transparently: a U.S. 510(k) compilation runs $15,000–$20,000, an EU technical-documentation or CER project is listed by class ($8,000–$30,000), a Canadian registration compilation is $3,000–$25,000 by class, and a regulatory-pathway determination is a flat $5,000. Every figure is quoted up front, never by the hour.
A worked example — one AI imaging algorithm, four markets. Take a single AI radiology tool already cleared by the FDA (Class II) and CE-marked (Class IIb) that you want to keep live across the United States, European Union, Brazil, and Singapore for a year. Pure Global's in-country representation totals $1,000 (US) + $2,000 (EU) + $3,000 (Brazil) + $3,000 (Singapore) = $9,000 for the year — flat, with every renewal, modification, and authority exchange included; you add one-time submission work only where a market actually requires a fresh dossier. That predictability is the point: when the rules differ in every market and your model keeps changing, the last thing a manufacturer needs is a regulatory bill that does too.
The convergence paradox
Here is the apparent good news. Beneath the fifteen-jurisdiction patchwork runs a powerful machinery of harmonization. IMDRF aligns the definitions and the Good Machine Learning Practice principles. The Medical Device Single Audit Program (MDSAP) lets a single quality-system audit satisfy five regulators at once — the U.S., Canada, Brazil, Japan, and Australia (FDA). And reliance pathways are spreading fast: Singapore accepts five reference agencies and routes ~98% of applications through abridged review; Brazil, Mexico, Australia, Malaysia, Vietnam, and the Gulf all recognize foreign approvals to some degree. In February 2026 IMDRF even published a global Reliance Playbook to codify the practice (IMDRF N89).
Which markets accept which foreign approvals (reliance routes)
An FDA clearance or CE mark is a master key to dozens of markets — each with its own lock.
| Market | Recognized reference agencies / programs | Effect |
|---|---|---|
| Singapore (HSA) | US FDA, EU NB, Health Canada, TGA, Japan MHLW | Abridged/expedited/immediate; ~98% eligible |
| Brazil (ANVISA) | TGA, Health Canada, US FDA, Japan MHLW (Class III/IV) | "Optimized analysis" ~20–30% faster |
| Mexico (COFEPRIS) | All IMDRF members + MDSAP participants | Abbreviated pathway, 30 working days |
| Australia (TGA) | US FDA, Health Canada, MHLW/PMDA, EU NB, MDSAP | Abridged conformity assessment |
| Malaysia (MDA) | US FDA, Health Canada, TGA, EU CE, PMDA, HSA, Thai FDA | Verification (abridged) route + MDSAP |
| Vietnam (MOH) | US FDA, EU, PMDA, TGA, Health Canada, MFDS, NMPA | Unusually broad SRA fast-track |
| Saudi Arabia (SFDA) | FDA/CE supportive only | Full technical-file review still required |
| UAE (EDE) | CE, US FDA | Reliance-based registration |
| MDSAP (one audit) | US, Canada, Brazil, Japan, Australia | Single QMS audit accepted by all five |
Source: HSA, ANVISA, COFEPRIS, TGA, MDA Malaysia, Thai FDA, Vietnam MOH, SFDA, EDE — Pure Global analysis, June 2026.
For a static device, this is transformative: one strong approval — typically FDA or CE — becomes a master key that opens dozens of markets at abridged speed and cost. This is precisely the leverage a well-run market-access program is built to exploit.
And here is the paradox. For adaptive AI, the master key stops working at exactly the point that matters most — change control. The convergence is on the device; the divergence is on the AI. Consider the same machine-learning model seeking to update its algorithm:
- In the U.S., a pre-authorized PCCP lets it ship the update with no new submission.
- In the EU, a "substantial" software change still triggers Notified Body re-review — and, layered on top, a separate AI Act conformity assessment.
- In China, the update is tolerated only if "the core algorithm remains unchanged"; a genuine retraining means full change registration.
- In Korea, the DMPA permits pre-approved change plans — but only within pre-approved parameters.
As one analysis puts it bluntly, "a PCCP authorized by the FDA does not satisfy EU AI Act obligations, and vice versa" (Berkley Lifesciences). No MDSAP audit and no reliance route resolves this. An AI developer who wins clearances in ten markets has not bought peace; they have bought ten different change-control obligations, each triggered every time the model improves. For a technology whose entire value proposition is that it keeps getting better, that is a structural, compounding tax on success — and it falls hardest on the small, fast-moving teams that build the best models.
The next frontier: generative AI and foundation models
If adaptive AI strained the system, generative AI threatens to overwhelm it. Everything above assumes a model trained for a single, well-defined intended use — detect a bleed, measure an ejection fraction, flag a nodule. Large language models and multi-modal foundation models break that assumption in three ways at once: they are general-purpose (one model, many possible uses), non-deterministic (the same prompt can yield different answers), and prone to hallucination (confident, fluent, wrong). None of those properties fits comfortably inside a framework built around a fixed intended use and a "locked" reference version.
Regulators know it. The WHO issued the first global guidance dedicated to large multi-modal models in January 2024, warning specifically about fabricated outputs, automation bias, and the difficulty of validating systems trained on internet-scale data (WHO). The FDA's Digital Health Advisory Committee devoted its inaugural meeting, in November 2024, to the total-product-lifecycle challenges of generative AI-enabled devices (FDA). And South Korea, characteristically first, published the world's first guideline for generative-AI medical devices in January 2025 and authorized its first such device in April 2026.
But guidance is not the same as a clearance pathway. As of mid-2026, the established routes — 510(k), De Novo, CE marking — still assume a device you can pin down, test against a fixed standard, and freeze. A general-purpose clinical LLM satisfies none of those preconditions cleanly, which is why the first wave of "generative AI in healthcare" has reached the market mostly as administrative tools (ambient scribes, documentation aids) that sidestep the device definition, rather than as cleared diagnostic devices. The regulatory frontier for autonomous, generative clinical AI is, genuinely, still being drawn — and the markets that draw it first (Korea today; others to follow) will shape how the rest of the world copies them. For developers, the practical lesson is to watch where the lines are being set and to design the regulatory strategy, not just the product, for a moving target.
The market-access playbook
If the problem is fragmentation, the answer is a system. Across the patterns above, a repeatable playbook emerges for getting an AI medical device to the world — and keeping it there.
1. Classify before you build the evidence. The same product can be Class II or Class III depending on the market and the claim. Map the target markets' classification rules first, because they dictate the clinical and technical evidence you will need. Sequencing the claim and the evidence to the strictest target market avoids rebuilding the dossier later.
2. Win a strong anchor approval — then exploit reliance deliberately. An FDA clearance or CE mark is worth far more than one market; it is the credential that unlocks abridged routes in Singapore, Brazil, Mexico, Australia, the Gulf, and beyond. The art is knowing which anchor each target market recognizes, and routing the dossier accordingly. Saudi Arabia, which demands local validation even of foreign-approved AI, is the reminder that reliance is a map, not a blanket.
3. Stand up in-country representation where it is mandatory — which is most places. A foreign manufacturer cannot hold its own registration in Brazil (BRH), the EU (Authorized Representative), China (legal agent), Japan (MAH/DMAH), Saudi Arabia, the UAE, India, and many more. Each requires a local legal entity to hold the registration and front the health authority. Standing up thirty such entities is impractical; outsourcing them to a single partner is how scale becomes feasible.
4. Treat change control as a first-class, multi-market workstream. This is the AI-specific discipline. Build a U.S. PCCP, but also map how each market handles the same update — and design the algorithm's release cadence around the most restrictive regime that matters to your business. The lifecycle plan is now part of the market-access plan.
5. Run it as one connected operation, not thirty disconnected filings. The cost is not any single registration; it is the coordination — translations, local holders, renewal calendars, change notifications, and post-market vigilance across dozens of regimes that drift independently.
A typical sequence in practice. For an imaging-AI developer, the rollout often runs: secure the FDA 510(k) or CE mark as the anchor; in parallel, file in your home market and one fast reliance market (Singapore or Australia) to bank early revenue; use those approvals to open the abridged routes in Brazil, Mexico, and the Gulf; then take on the high-effort, high-value markets — China and Japan — where local testing or clinical data is unavoidable and the lead times are longest. Throughout, a single change-control plan is maintained centrally and mapped to each market's update rules, so a model improvement is filed everywhere it must be, and nowhere it needn't be. The order is not arbitrary; it sequences cash flow, evidence reuse, and the scarce-resource markets deliberately.
This is the work Pure Global is built to do: in-country representation and AI-assisted regulatory execution across 30+ markets, delivered on a flat annual fee rather than the open-ended hourly model the industry defaults to. The data in this report — drawn from the FDA, EUDAMED, NMPA, PMDA, MFDS, ANVISA and dozens of national registries, alongside our own market-by-market cost and timeline dataset — is the same intelligence we use to sequence a client's global rollout. The point of mapping the maze this thoroughly is to be able to walk a client through it quickly.
Conclusion: four things to take away
AI is now a mainstream device category, and the data proves it. From roughly 1 in 700 FDA clearances in 2019 to 1 in 28 in 2025, with 1,500-plus AI devices authorized in the U.S. alone and national programs in Korea, China, Taiwan, and Japan scaling fast, AI SaMD has crossed from novelty to norm.
Every regulator up-classifies it, and most are converging on lifecycle oversight. Rule 11 in the EU and Brazil, Class III in China, high-risk under the EU AI Act — diagnostic and therapeutic AI is treated as serious, and the shared answer is total-product-lifecycle control, GMLP, and predetermined change plans.
Reliance makes one approval travel — except for the AI part. The harmonization machinery (IMDRF, MDSAP, reliance routes) genuinely lets a strong anchor approval unlock dozens of markets. But adaptive-AI change control diverges sharply, so a single approval does not stay valid everywhere as the model evolves. That gap is the defining operational challenge of the field.
The competitive edge is the registration machine, not the registration. When the rules differ in every market and change every year, the durable advantage belongs to those who can register fast, everywhere, and keep each approval alive through every model update — as one connected system.
Talk to us
If you are building or scaling an AI medical device and weighing which markets to enter, in what order, and how to keep each approval valid as your model improves, that is exactly the problem we solve. Talk to Pure Global about a market-access plan built on the data above — or explore our market-by-market registration guides to go deeper on any single country.
Sources
Government & harmonization bodies cited inline above; key references grouped below. All figures are dated; market-size and forecast figures are third-party estimates whose scope varies and should be read as ranges.
Definitions, frameworks & guiding principles
- IMDRF — SaMD: Key Definitions (N10, 2013); Risk Categorization (N12, 2014); ML-enabled Medical Devices: Key Terms (N67, 2022); GMLP Guiding Principles (N88, 2025); Reliance Playbook (N89, 2026). imdrf.org
- US FDA — Software as a Medical Device (SaMD); Proposed Regulatory Framework for Modifications to AI/ML-Based SaMD (2019); AI/ML Action Plan (2021). fda.gov
- WHO — Ethics & governance of AI for health (2021); Regulatory considerations on AI for health (2023); LMM guidance (2024); Good Reliance Practices, TRS 1033 Annex 10 (2021). who.int
United States
- FDA — Artificial Intelligence-Enabled Medical Devices list (Q1-2026 update, 1,524 devices; ~76% radiology); PCCP final guidance (Dec 2024); Digital Health Advisory Committee on generative AI (Nov 2024); MDUFA FY2026 fees. fda.gov · The Imaging Wire (radiology-share analysis, 2026); Innolitics & IntuitionLabs (clearance trackers, 2025–26).
European Union & UK
- EUR-Lex — Regulation (EU) 2017/745 (MDR), Annex VIII Rule 11; Regulation (EU) 2024/1689 (AI Act), Articles 6, 99, 113. European Commission — MDCG 2019-11 Rev.1 & MDCG 2025-6. MedTech Europe & Team-NB (Notified Body capacity). MHRA — Software & AI Change Programme; AI Airlock; CE-recognition guidance. gov.uk
Canada, Australia, Japan, Korea
- Health Canada — Pre-market guidance for ML-enabled medical devices (Apr 2026). canada.ca
- TGA — software reforms (2021); AI consultation outcomes (2024–26). tga.gov.au
- PMDA/MHLW — SaMD guidance; DASH for SaMD; IDATEN. pmda.go.jp
- MFDS — 2025 Approval Report (153 AI devices); DMPA; generative-AI guideline. mfds.go.kr; bioin.or.kr
Asia-Pacific, LatAm, MEA
- HSA Singapore — GL-04-R4; reliance/reference agencies. hsa.gov.sg; moh.gov.sg
- NMPA China — AI classification & Oct 2025 reform. nmpa.gov.cn · JMIR Medical Informatics (154 AIMDs, 2026).
- CDSCO India — Draft MD Software guidance (Oct 2025). TFDA Taiwan — CADe/CADx guidance; J. Formos. Med. Assoc. (166 licenses).
- ANVISA Brazil — RDC 751/2022 (Rule 11), RDC 657/2022, IN 290/2024. gov.br/anvisa
- COFEPRIS Mexico — Abbreviated Pathway (2025). SFDA Saudi Arabia — MDS-G010 (2022). UAE EDE — Federal Decree-Law 38/2024. SAHPRA South Africa — AI communication (2025).
Market, companies, patents, safety
- Market size: Grand View Research; MarketsandMarkets; The Business Research Company; Mordor Intelligence; Fortune Business Insights (2025–26).
- Funding & companies: Rock Health (2025 digital-health funding); CB Insights; company FY2025 filings (Tempus AI, iRhythm, HeartFlow, Butterfly Network, Lunit, VUNO); Aidoc (Series E).
- Patents: WIPO Technology Trends (2019) & Generative AI Patent Landscape (2024); CNIPA.
- Safety: NEJM (pulse-oximeter bias, 2020); JAMA Internal Medicine (Epic Sepsis Model, 2021); JAMA Health Forum & JAMA Network Open (AI recalls, 2025); FDA pulse-oximeter guidance.
- Trade: OECD-IMF-WTO Measuring Digital Trade (2021); WTO; UNCTAD.
- Pure Global proprietary market-access cost & timeline dataset (2026); openFDA, EUDAMED & MFDS database analysis — Pure Global, June 2026.
Latest Blog Content
Explore our collection of articles, success stories, and regulatory updates, designed to help you take your product global.



Let's Talk,
Anywhere You Are.
Whether looking for more information or ready to partner with us, we're here to guide you through every step of the regulatory process.
Contact us