
AI come dispositivo medico: la mappa globale del regolamento, registrazione e accesso al mercato
L'intelligenza artificiale è la classe più rapida crescita di dispositivi medici nella storia — e la più regolazione-divergenza. La FDA degli Stati Uniti ora elenca 1.524 dispositivi AI-enabled; Corea del Sud ha autorizzato 153 in un solo anno. Eppure lo stesso software è Classe II negli Stati Uniti, Classe IIa+ e "alto rischio" nell'UE, e Classe III in Cina — ciascuno con le proprie prove, azionisti locali e regole di controllo dei cambiamenti. Questo rapporto mappa come ogni regolatore principale classifica, approva e polizia AI come un dispositivo medico, con i costi di registrazione, le linee temporali e le rotte di dipendenza attraverso 30+ mercati, e il playbook per raggiungerli senza ricostruire il dossier ogni volta.
Una guida sul campo basata su prove su come i regolatori del mondo classificano, approvano e polizia intelligenza artificiale nel settore sanitario - e come raggiungere 30+ mercati senza ricostruire il dossier ogni volta.
TL; RD
L'intelligenza artificiale è diventata la categoria più rapida di dispositivi medici nella storia. L'elenco pubblico della Food and Drug Administration negli Stati Uniti dei dispositivi abilitati all'AI ha raggiunto le autorizzazioni 1,524 entro la metà del 2026, con circa tre quarti in radiologia (FDA). Sulla nostra analisi del database FDA 510(k), la parte di AI di tutte le autorizzazioni scalate da circa 1 nel 700 nel 2019 a circa 1 in 28 nel 2025. Solo la Corea del Sud ha autorizzato i dispositivi AI nel 2025 (MFDS).
Ma lo stesso software che naviga attraverso un'agenzia può stallo nel prossimo. Ecco l'argomento di questa relazione, in cinque punti:
- AI è un dispositivo, e la maggior parte di esso è "Software come dispositivo medico" (SaMD Quando il software diagnostica, triages, o raccomanda il trattamento, è regolato come un bisturi o uno scanner — in decine di giurisdizioni, ciascuno con le proprie regole.
- Il mondo intero lo riclassifica. La regola MDR dell'UE 11, la regola 11 del Brasile, la classe III della Cina di default per AI diagnostico — il software diagnostico e terapeutico quasi ovunque atterra in una classe di rischio superiore che richiede la revisione di terze parti.
- L'AI adattiva ha infranto il modello di approvazione una volta. Un modello che continua a imparare i cambiamenti dopo che le navi, così i regolatori hanno inventato nuovi meccanismi — il piano di controllo predeterminato degli Stati Uniti, IDATEN del Giappone, DMPA della Corea — che non non corrispondono a vicenda.
- Il rispetto converge, i divergenti dell'AI. Una rete di percorsi di affidamento e riconoscimento dovrebbe permettere a un'approvazione di sbloccare molti mercati. Funziona — per dispositivi statici. Per adaptive AI, "un PCCP autorizzato dalla FDA non soddisfa gli obblighi dell'UE AI Act e viceversa" (Berkley Lifesciences).
- Così i vincitori industrializzano la registrazione multi-mercato. Il bordo competitivo non è più un'unica autorizzazione; è la macchina operativa che trasforma un'approvazione in trenta, e mantiene ciascuno valido come l'algoritmo si evolve.
Questo è il divario Pure Global chiude — la rappresentazione in-paese e l'esecuzione regolamentare assistita dall'AI su 30+ mercati, su una tassa annuale piatta. Il resto della relazione è la mappa.
Che cosa "AI come dispositivo medico" in realtà è
Inizia con la parola che fa il sollevamento pesante: dispositivo. Se un pezzo di software è "destinato per l'uso nella diagnosi, nel trattamento o nella prevenzione della malattia", è un dispositivo medico in quasi ogni sistema legale sulla terra — se funziona su un chip all'interno di uno scanner MRI o come app nel browser di un radiologo. L'IA non ottiene il proprio statuto nella maggior parte dei paesi; eredita l'intero apparato di diritto medico-dispositivo.
La definizione di ancoraggio proviene dal International Medical Device Regulators Forum (IMDRF), il corpo in cui le principali agenzie armonizzano il loro vocabolario. Nel suo documento di fondazione 2013, IMDRF ha definito Software come dispositivo medico (SaMD) come "software destinato ad essere utilizzato per uno o più scopi medici che eseguono questi scopi senza far parte di un dispositivo medico hardware" (IMDRF N10). La FDA ha adottato quella lingua verbatim e disegna una distinzione a tre vie che conta per tutto a valle (FDA):
- SaMD — software che *è il dispositivo medico (un algoritmo di triage toracico-X-ray, un rilevatore di diabetico-retinopatia). Qui vive la maggior parte dell'AI clinica.
- SiMD — software in un dispositivo medico, integrale all'hardware (il firmware che esegue una pompa di infusione).
- Software usato per fabbricazione o mantenere un dispositivo, che è regolato in modo diverso di nuovo.
Per l'IA specificatamente, il documento di key-terms 2022 di IMDRF definisce un Machine Learning-enabled Medical Device come "un dispositivo medico che utilizza l'apprendimento automatico, in parte o in tutto, per raggiungere il suo scopo medico previsto" (IMDRF N67).
La distinzione che rompe tutto: bloccato vs. adattativo
La regolazione tradizionale del dispositivo poggia su un semplice affare: si prova che un dispositivo è sicuro ed efficace once, a un design fisso, e che il design poi rimane messo. L'intelligenza infrange quell'ipotesi. La carta di discussione della FDA 2019 ha disegnato la linea con precisione. Un "locked" algoritmo è uno che "fornisce lo stesso risultato ogni volta che lo stesso input viene applicato e non cambia con l'uso" — una tabella di ricerca, un albero di decisione, un classificatore congelato (FDA, 2019). Un adaptive o un algoritmo di apprendimento continuo cambia dopo che viene distribuito.
Questa singola proprietà — software che si migliora nel campo — è la ragione per cui AI ha bisogno di un decennio di nuovo regolamento. Se il prodotto approvato a gennaio non è il prodotto in esecuzione in un ospedale a giugno, che cosa esattamente ha approvato? Ogni quadro di questa relazione è, in fondo, un tentativo di rispondere a questa domanda, e il Total Product Life Cycle (TPLC) — la supervisione di tutta la vita del dispositivo, non solo il momento della clearance — è la risposta condivisa.
Come i regolatori decidono quanto difficile guardare
Il framework di risk-categorizzazione 2014 di IMDRF ha definito la logica che il mondo segue:SaMD merita una scala con due fattori: il significato delle informazioni che fornisce (lo fa informa, drive, o diagnose/trattamento) e la gravità della situazione sanitaria (non grave, grave o critica) (IMDRF N12). Un'applicazione che suggerisce tratti per il mal di schiena e un algoritmo che segnala un sanguinamento cerebrale non sono lo stesso animale regolamentare, e questa griglia a due assi è il motivo.
La Organizzazione Mondiale della Sanità ha aggiunto il ponteggio etico. La sua relazione del 2021 L'etica e la governance dell'intelligenza artificiale per la salute stabiliscono sei principi — proteggono l'autonomia; promuovono il benessere e la sicurezza; garantiscono trasparenza e spiegabilità; adottino responsabilità e responsabilità; garantiscano l'inclusione e l'equità; e promuovano l'intelligenza artificiale sostenibile (AI) WHO). L'OMS ha seguito con considerazioni di regolamentazione nel 2023 e, nel gennaio 2024, la prima guida globale mirata in maniera quadrata all'AI generativa e ai grandi modelli multimodali (WHO).
Come siamo arrivati
I dispositivi medici dell'AI non sono arrivati con una sola svolta; si accresero, paese per paese, oltre un decennio. Le pietre miliari sottostanti tracciano come una definizione nel 2013 è diventata regole del ciclo di vita dedicate in cinque continenti.
Una linea temporale globale della regolazione AI medical-device (2013–2026)
In appena un decennio, AI SaMD è passata da una definizione IMDRF a regole del ciclo di vita dedicate in cinque continenti.
| Data | Milestone |
|---|---|
| Dicembre 2013 | IMDRF N10 definisce "Software come dispositivo medico" (SaMD) |
| Gennaio 2017 | FDA cancella Arterys — prima nuvola + strumento clinico deep-learning (510(k)) |
| Apr 2018 | FDA autorizza IDx-DR — prima diagnostica AI autonoma (De Novo) |
| Maggio 2018 | Corea MFDS approva VUNO Med-BoneAge — il primo dispositivo AI della Corea |
| Dicembre 2018 | Giappone PMDA approva EndoBRAIN (classe III) — la prima AI del Giappone SaMD |
| Apr 2019 | Carta di discussione FDA sulle modifiche a AI/ML SaMD(locked vs adattativo) |
| 2020 | Cina NMPA approva DeepVessel FFR — dispositivo AI di prima classe III; Giappone lancia IDATEN + DASH per SaMD |
| Gennaio 2021 | FDA AI/ML Piano d'azione; Jun 2021 |
| Ottobre 2021 | GMLP — 10 principi guida (FDA + Health Canada + UK MHRA) |
| 2022 | Saudi SFDA MDS-G010 — guida per dispositivi AI (citata da alcuni come il primo "applicabile"); termini IMDRF N67 ML; Brasile RDC 751/657 |
| 2024 | Entra in vigore la legge UE sull'AI; orientamento LMM (generativo-AI) (Gran 2024) |
| 2024 | FDA finalizza Predetermined Change Control Plan (PCCP) guida |
| Gennaio 2025 | IMDRF N88 GMLP finale; Corea emette la prima linea di guida del dispositivo generativo-AI del mondo; FDA AI lifecycle bozza di guida |
| 2026 | IMDRF N89 Reliance Playbook |
| Apr 2026 | Health Canada finalizza la guida ML-device; Corea approva il primo dispositivo generativo-AI |
Fonte: compilato da IMDRF, US FDA, EU, NMPA, PMDA, MFDS, SFDA, ANVISA, Health Canada e documenti primari dell'OMS —Pure Global, giugno 2026.
Alcuni momenti meritano l'enfasi. In gennaio 2017, la FDA ha eliminato Arterys - il primo strumento clinico per combinare cloud computing e deep learning (PR Newswire). Poi, in April 2018, è venuto il watershed: la FDA ha autorizzato IDx-DR, la prima AI ha permesso di fornire una diagnosi autonomamente, senza medico interpretare il risultato — uno schermo diabetico-retinopatia per la cura primaria che ha colpito la sensibilità dell'87,2% e la specificità del 90,7% nel suo processo cardine (Medicina digitale npj). Entro mesi, la Corea (VUNO Med-BoneAge, maggio 2018) e il Giappone (EndoBRAIN, dicembre 2018) hanno approvato i loro primi; La Cina ha seguito nel 2020 con DeepVessel FFR, il suo primo dispositivo AI di classe III (Medicina digitale npj).
Il ponteggio regolamentare catturato tra il 2021 e il 2025: il trilaterale Good Machine Learning Practice principi nell'ottobre 2021, l'UE AI Act entrare in vigore nell'agosto 2024, l'ultima Predetermined Change Control Plan guida nel dicembre 2024, e — dicendo quanto velocemente la frontiera si muove — Corea prima linea generativa-AI dispositivo BioWorld). Entro febbraio 2026, IMDRF aveva pubblicato un Reliance Playbook globale per aiutare i regolatori a appoggiarsi sul lavoro dell'altro (IMDRF N89).
I dati: quanto grande, quanto veloce, dove
La curva di sdoganamento si piega bruscamente verso l'alto
Il miglior indicatore della penetrazione di AI in medicina è il pubblico della FDA Artificial Intelligence-Enabled Medical Device s lista, che ha raggiunto 1,524 autorizzazioni sul suo aggiornamento più recente (riflettendo i dati attraverso il primo trimestre del 2026) (FDA). Per vedere la velocity, abbiamo analizzato il database sottostante FDA 510(k) direttamente. I titoli Identificati AI/ML sono passati da una manciata nel tardo 2010 a 115 nel 2025 — e, più chiaramente, l'IA è cresciuta da 0,14% di tutte le autorizzazioni 510(k) nel 2019 al 3,57% nel 2025, un salto di circa 25 volte in quota in sei anni.
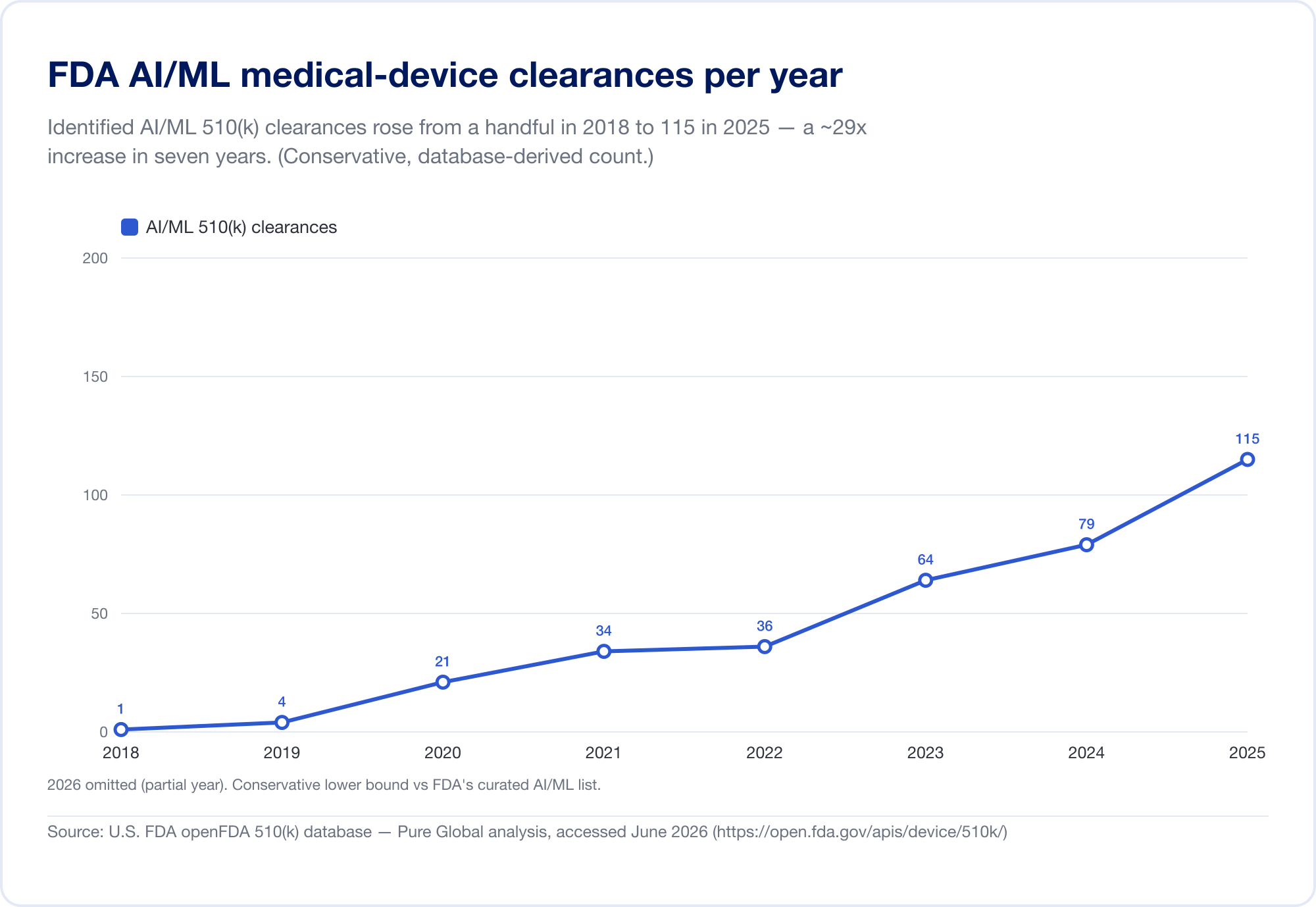
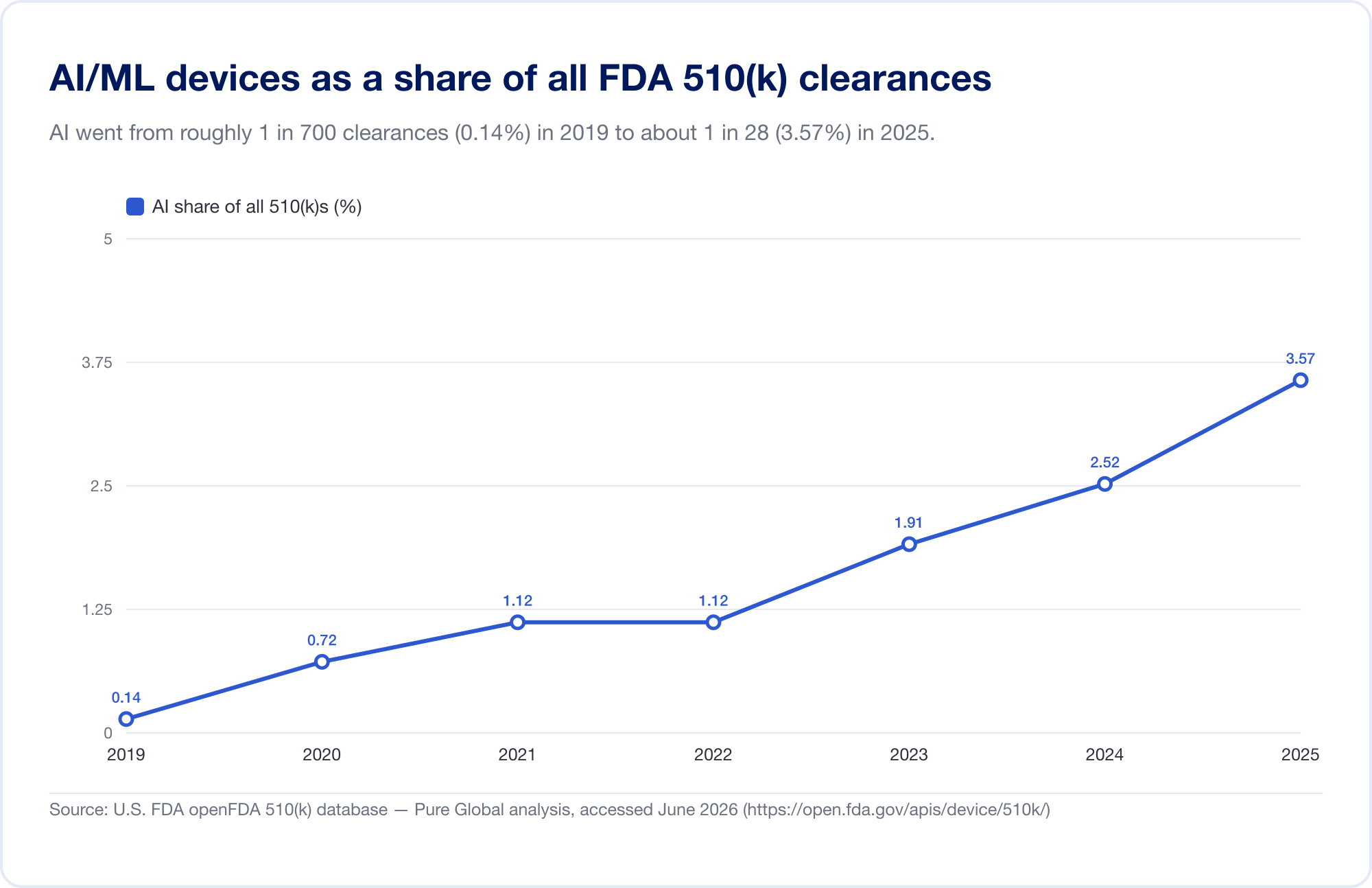
(Il nostro database-derivato conteggio è volutamente conservatore — più stretto della lista curata della FDA, perché molti strumenti di radiologia AI siedono sotto i codici di prodotto il cui testo non dice mai "AI". Lo usiamo per tendenza e geografia; la lista della FDA è il totale dei titoli. Fonte: open FDA 510(k) database —Pure Global analisi, giugno 2026.)
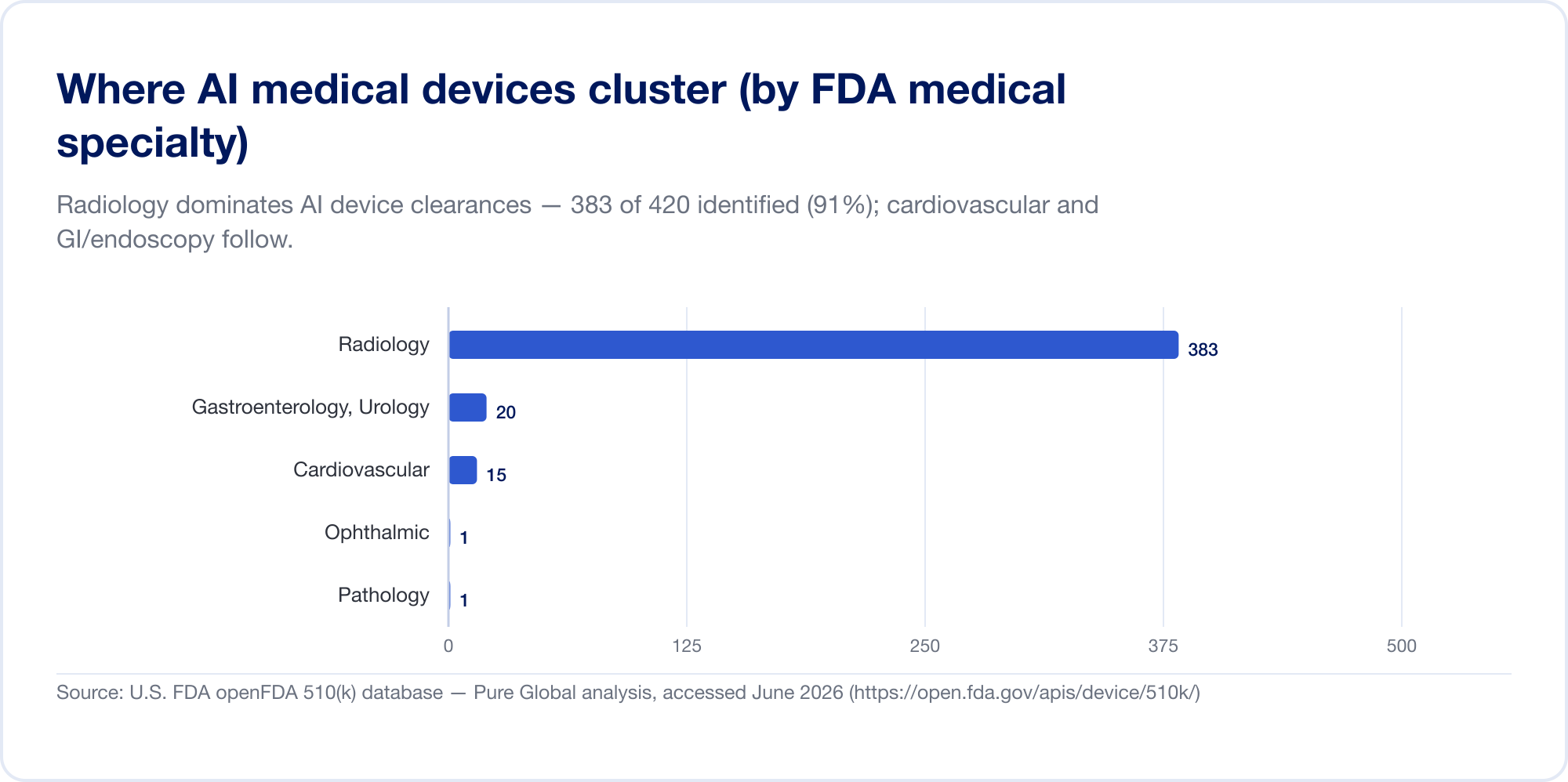
È travolgente una storia di imaging — per ora
L'IA in medicina è, oggi, per lo più AI in radiologia. Sulla lista della FDA, la radiologia rappresenta circa il 76% di tutte le autorizzazioni AI (Il filo Imaging); nel nostro campione di clearance la concentrazione è ancora più alta, con cardiovascolare e gastroenterologia (endoscopia) un secondo e terzo lontano. Il motivo è strutturale: l'imaging è digitale, abbondante e etichettato, e il percorso 510(k) permette a un nuovo algoritmo di citare uno esistente come un predicato. La patologia, i segnali cardiologici e i modelli clinici-text stanno crescendo, ma il centro di gravità rimane l'immagine.
Gli innovatori sono ovunque; i mercati sono ovunque
Ecco il fatto che dovrebbe ridefinire qualsiasi piano di commercializzazione. I dispositivi medici dell'AI liberati negli Stati Uniti provengono da almeno 16 paesi. Nella nostra analisi di AI 510(k) candidati, gli Stati Uniti guida, ma Corea del Sud, Israele, Canada, Francia, Cina, Germania, Regno Unito, Paesi Bassi, Taiwan e Giappone ogni campo numeri significativi.
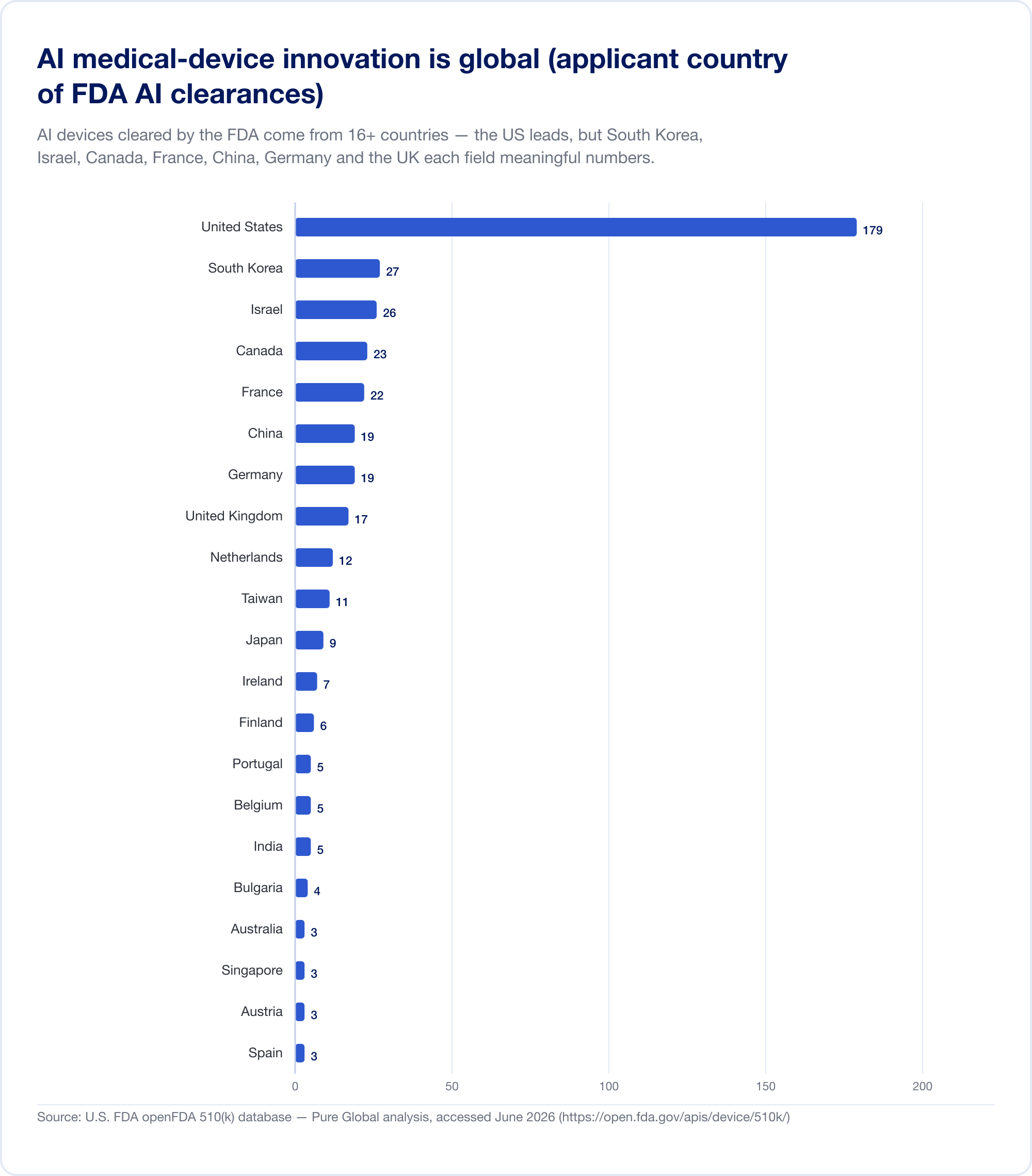
L'uscita nazionale conferma il modello. Corea del Sud autorizzato (approvazioni e certificazioni combinate) 64 dispositivi AI nel 2023, 108 nel 2024, e 153 nel 2025 — un salto del 41,6%, con 77,7% fatti internamente (MFDS La Cina aveva approvato circa 154 dispositivi medici AI entro la metà del 2025, circa l'80% di essi nella classe III ad alto rischio (JMIR Informatica medica Taiwan con licenza 166 dispositivi AI/ML tra il 2020 e il 2024 (J. Formos. Med. Assoc. Japan aveva 51 SaMD sulla lista PMDA del 2025 settembre (Salute e Medicina Globale).
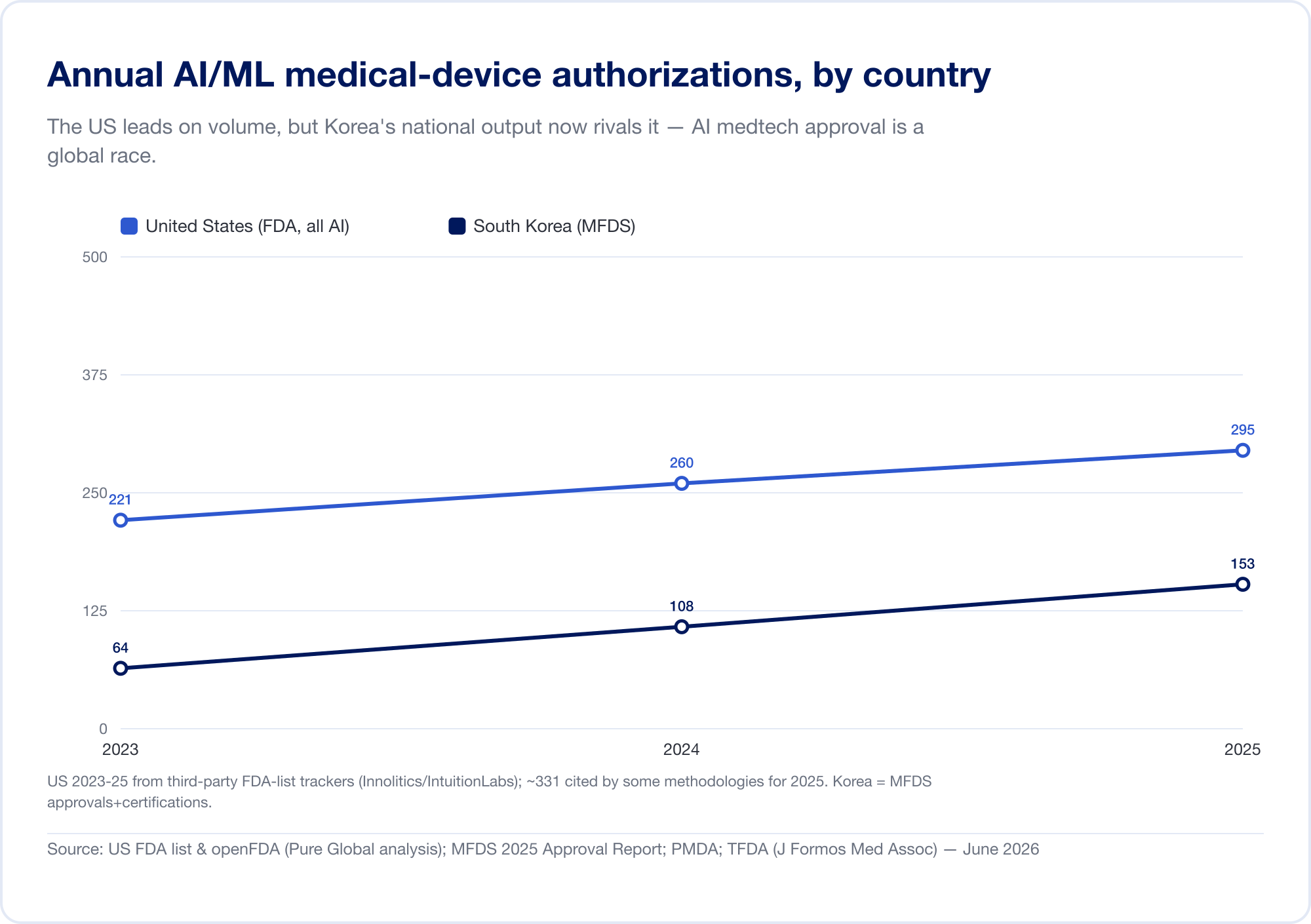
L'implicazione è diretta: un brillante algoritmo costruito a Tel Aviv, Seoul, o Shanghai deve raggiungere i pazienti nei mercati che ciascuno parla un linguaggio normativo diverso. L'innovazione è globale; l'approvazione è ostinatamente locale.
Quanto è grande il mercato? Dipende interamente da ciò che conta
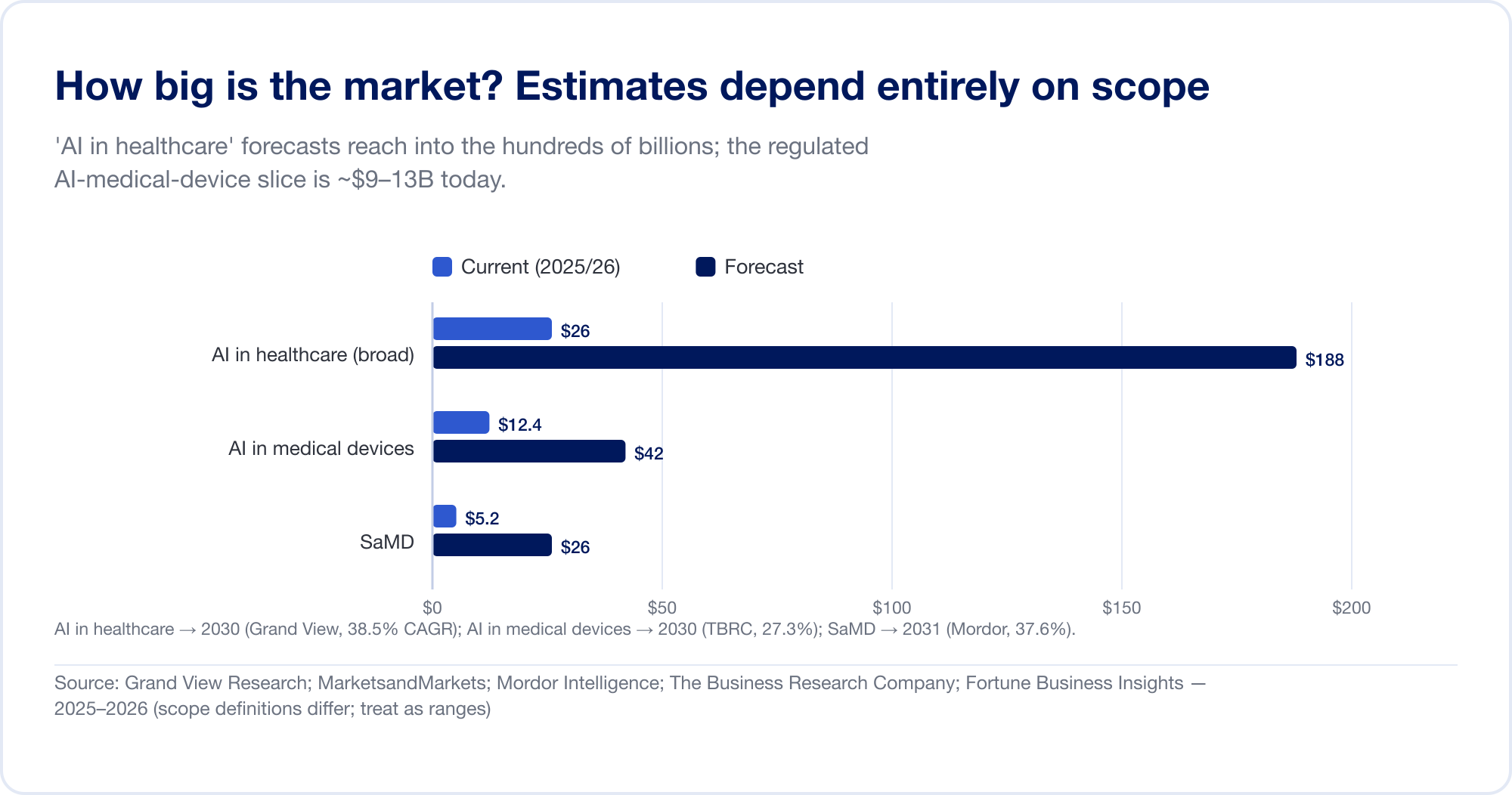
Le stime di mercato per l'IA nel settore sanitario abbracciano un ordine di grandezza, perché gli analisti disegnano il confine in luoghi selvaggiamente diversi. L'ampio "AI nel settore sanitario" categoria — che raggruppa la scoperta della droga, l'automazione amministrativa, gli scribi ambientali, e più — è previsto a USD 187,7 miliardi entro il 2030 ad un CAGR del 38,5% (Grand View Ricerca), con la casa più aggressiva che proietta oltre USD 1 trilioni entro il 2034 (Fortune Business Insights). La fetta più stretta e regolamentata — AI in dispositivi medici — è molto più piccola e più credibile: circa USD 12,4 miliardi nel 2025, raggiungendo USD 42,4 miliardi entro il 2030 (Azienda di ricerca aziendale). Puro SaMD è stimato a circa 5–25 miliardi di USD a seconda della fonte (Intelligenza di Mordor). Tratta ciascuno di questi come una gamma, non un fatto.
La capitale racconta una storia più pulita. U.S. finanziamento di impresa digitale-sanitaria rimbalzato a USD 14,2 miliardi nel 2025 (+35%), e per la prima volta le startup abilitate all'AI hanno catturato la maggioranza — 54% di tutti i dollari (Rock Salute). AI pubblico-SaMD i nomi sono inflessibili dalla crescita alla redditività: Tempus AI ha pubblicato 1,3 miliardi di dollari in ricavi FY2025 (+83%) (Tempus); iRhythm ha raggiunto 747 milioni di USD e il suo primo trimestre GAAP-profit (IRhythm HeartFlow è diventato pubblico nell'agosto del 2025 ed è cresciuto del 40% a 176 milioni di USD (HeartFlow); la Corea Lunit è cresciuta del 53% con il 92% dei ricavi da oltreoceano (Lunit). Tra i giocatori privati, Aidoc ha sollevato una serie E di 150 milioni di dollari nel mese di aprile 2026, prendendo il finanziamento totale oltre mezzo miliardo di dollari (Aidoc).
La gara dei brevetti ha un leader diverso
Proprietà intellettuale è dove il concorso globale è più visibile. Lo studio di riferimento di WIPO ha contato circa 340.000 brevetti relativi all'intelligenza artificiale, con la vita e le scienze mediche il terzo campo di applicazione più grande (WIPO). Ma la geografia è da allora capovolta: nel decennio al 2023, La Cina ha presentato 38.210 invenzioni generative-AI — più del resto del mondo combinato — contro 6.276 dagli Stati Uniti (WIPO). L'ufficio brevetti della Cina ha rilasciato le linee guida dedicate all'esame dei brevetti AI l'ultimo giorno del 2024 (CNIPA). Il gasdotto dell'IA medica viene riempito in modo sproporzionato dall'Asia — che rende la strategia di accesso multi-mercato una questione di * when*, non if, per una quota crescente degli sviluppatori del mondo.
Il punto cieco: non potete vedere SaMD in dati commerciali
Una cautela analitica che ridefinisce come pensare a questo mercato. L'intelligence del mercato medtech tradizionale si basa su dati di importazione e doganali, ma pure SaMD è invisibile ad esso. Il software fornito da cloud download o app store non attraversa alcun confine fisico, non genera alcuna dichiarazione doganale HS-code, e non viene catturato nelle statistiche di commercio merce. Gli standard internazionali dicono così direttamente: "I flussi di dati non direttamente monetizzati non sono generalmente considerati come flussi commerciali negli attuali standard statistici" (OCSE-IMF-WTO), e la moratoria dell'OMC sui dazi doganali per le trasmissioni elettroniche si è tenuta dal 1998 (UNCTAD). AI-embedded hardware — un AI-enabled CT scanner — si muove come un bene e si presenta nei dati; un algoritmo cloud 510(k)-cleared non lo fa. L'upshot: analisi commerciale convenzionale sistematicamente under-counts software AI e hardware over-weights. L'unica impronta affidabile di un SaMD«la diffusione globale è la sua registrazioni — che è esattamente l'obiettivo che questa relazione utilizza.
Il calcolo della sicurezza
Dietro ogni serraggio delle regole è un corpo di prova che l'IA in medicina può fallire in modi che i dispositivi tradizionali non lo fanno. Tre risultati, in particolare, rimodellano come i regolatori pensano.
Bias che si nasconde nei dati normali. Un punto di riferimento New England Journal of Medicine studio ha scoperto che gli ossimetri del polso — onnipresenti e sempre più accoppiati con algoritmi — hanno perso livelli pericolosamente bassi di ossigeno nel sangue (ipoxemia occupata) nel 117% dei pazienti neri rispetto al 3,6% dei pazienti bianchi, una disparità approssimativamente triplice in circa 50.000 letture accoppiate (NEJM, 2020). La FDA ha rilasciato una comunicazione di sicurezza nel febbraio 2021 e, entro il gennaio 2025, bozza di guida esigente più diversificata validazione attraverso toni della pelle (FDA). La lezione generalizzata: un modello formato su una popolazione non rappresentativa può portare quel bias silenziosamente in ogni ospedale che lo dispiega.
Valida che non sopravvive al contatto con la realtà. Il modello Epic Sepsis — uno strumento di previsione proprietario distribuito a centinaia di ospedali statunitensi — è stato convalidato esternamente su 38.455 ospedalizzazioni e ha segnato un'area sotto la curva di 0.63, ben al di sotto della rivendicazione del venditore 0.76–0.83. Ha perso il 67% dei casi di sepsi durante il licenziamento avvisi il 18% di tutti i pazienti (JAMA Medicina interna, 2021). Un modello può essere "in produzione" in scala e ancora non funziona come pubblicizzato.
Richiama il cluster subito dopo l'autorizzazione. A 2025 Johns Hopkins/Yale analisi di dispositivi AI autorizzati dalla FDA ha scoperto che 43,4% dei richiamamenti AI-device si è verificato entro i primi 12 mesi di clearance — approssimativamente raddoppiare il tasso per 510(k) dispositivi complessivi (JAMA Health Forum, 2025). Uno studio parallelo trovato ricorda concentrati tra i dispositivi che non hanno pubblicato studi clinici (Rete JAMA aperta, 2025). Questioni di contesto: approssimativamente 97% dei dispositivi AI/ML chiaro attraverso il percorso 510(k), che non richiede prove umane prospettive - quindi un grande vantaggio sulla vigilanza post-mercato.
Il calcolo della sicurezza: perché i regolatori stanno stringendo su AI
Bias, gap di convalida e richiami anticipati sono le prove dietro il passaggio alla supervisione del ciclo di vita.
| Trovare | Figura | Fonte |
|---|---|---|
| Ipoxemia occulta dell'ossimetro, pazienti neri e bianchi | 11,7% vs 3,6% (~3x) | NEJM, dicembre 2020 |
| Epic Sepsis Modello AUC esterno (vs 0.76–0.83 rivendicato) | 0.63; 67% dei casi di sepsi | JAMA Intern. Med., giugno 2021 |
| AI-dispositivo richiama entro 12 mesi di distanza | 43,4% (~2x tutti i 510(k)s) | JAMA Health Forum, 2025 |
| Dispositivi AI richiamati (903 studiati) | 4,8%, concentrato in quelli mancanti studi clinici | Rete JAMA aperta, 2025 |
| Dispositivi AI ripuliti tramite 510(k) (nessun test prospettico richiesto) | ~97% | Analisi della lista FDA, 2025 |
Aggiungete a questo il problema di dataset shift — i modelli schierati si degradano silenziosamente come la popolazione del paziente, gli scanner o i sistemi di codifica intorno a loro cambiano — e avete la ragione per l'intero apparato moderno: Good Machine Learning Practice, Predetermined Change Control Plans, e il monitoraggio obbligatorio delle prestazioni del mondo reale. I regolatori non stanno stringendo perché l'intelligenza artificiale non funziona. Essi stanno stringendo perché funziona fino a quando tranquillamente non lo fa.
La mappa regolamentare globale
Questo è il nucleo di riferimento del rapporto: come le principali giurisdizioni effettivamente classificare, rivedere e polizia AI SaMD a metà del 2026. La linea d'osservazione è classificazione (che la classe di rischio atterra il software) e controllo di cambiamento (cosa succede quando l'algoritmo aggiorna). Una matrice di confronto consolidata segue i dettagli regionali.
Come 15 giurisdizioni regolano l'IA come dispositivo medico (2026)
*Same software, quindici risposte: classificazione, guida specifica dell'AI e regole di controllo del cambiamento divergono mercato per mercato *
| Competenza | Dove la maggior parte dell'AI SaMD terre | AI dedicato/SaMD orientamento | Cambio di controllo / meccanismo adattativo-AI | Affidabilità all'approvazione straniera |
|---|---|---|---|---|
| Stati Uniti (FDA) | Classe II (510(k)/De Novo) | Sì — PCCP finale 2024; progetto del ciclo di vita 2025 | PCCP (pre-autorizzazione modifiche, nessuna nuova presentazione) | No (rivista personale; basato sui predicati) |
| Unione europea | Classe IIa+ (Regola MDR 11) + AI Act ad alto rischio | MDR + AI Act + MDCG 2025- 6 | Sostanziale cambiamento →Notified Body revisione + AI Act | No (valutazione della conformità CE) |
| Regno Unito (MHRA) | Classe IIa+ (UK MDR 2002) | Software & AI Change Programme; AI Airlock | PCCP pianificato (strumento scenico) | CE accettato in GB a 2028/2030 |
| Canada (Health Canada) | Classe II-IV | Si — Guida ML finale apr 2026 | PCCP | MDSAP per QMS; non completa affidabilità del prodotto |
| Australia (TGA) | Classe IIa III | AI recensione 2024 (14 risultati) | Sviluppo | Itinerario comparabile-overseas-regolatore |
| Giappone (PMDA/MHLW) | Classe II-III | DASH per SaMD;SaMD orientamento | IDATEN (PACMP) modifiche pre concordate | Dati clinici stranieri accettati; MDSAP |
| Corea del Sud (MFDS) | Grado 2-3 | Si — incl. linea guida generativa-AI mondiale | DMPA piani di cambiamento pre-approvati | Limitato; propria recensione |
| Singapore (HSA) | Classe B–D | Sì — GL-04-R4 (2025), ciclo di vita AI-MD | Cambiare la notifica | Sì — 5 agenzie di riferimento (~98% abridged) |
| Cina (NMPA) | Classe III (software di decisione) | Sì — Principi CMDE AI + catalogo di classificazione | Carve-out solo se l'algoritmo di nucleo invariato | No (in paese agente; test di tipo) |
| India (CDSCO) | Classe A–D | Progetto unico (Otto 2025) | Protocollo di cambiamento algoritmo (proposto) | L'approvazione del paese facilita la Classe C/D |
| Taiwan (TFDA) | Classe II-III | Sì — guida CADe/CADx + PCCP | Guida PCCP (2024) | Valutazione delle prestazioni locali |
| Brasile (ANVISA) | Classe II-IV (Rule 11) | RDC 657 (SaMD); nessuna regola AI dedicata | Registrazione completa delle modifiche | Sì — IN 290/2024 (Classe III/IV, 4 agenzie) |
| Messico (COFEPRIS) | Classe I–III | Regole generali dei dispositivi | Registrazione | Sì — percorso abbreviato (IMDRF + MDSAP) |
| Arabia Saudita (SFDA) | Classe A–D | Sì — MDS-G010 (prima; citata come prima "applicabile") | Modifica della notifica tramite GHAD | Solo supporti; richiesta di validazione locale |
| UAE (EDE) | Classe I–III | Regole generali dei dispositivi | Registrazione | Sì — riconosce CE/FDA |
Fonte: US FDA, EU MDR/AI Act, MHRA, Health Canada, TGA, PMDA/MHLW, MFDS, HSA, NMPA, CDSCO, TFDA, ANVISA, COFEPRIS, SFDA, EDE —Pure Global analisi, giugno 2026.
Stati Uniti — il punto di riferimento e il più affollato
Gli Stati Uniti gestiscono il regime di dispositivi AI più attivo al mondo, amministrato dal Centro FDA per dispositivi e salute radiologica e dal suo Centro di Eccellenza Digitale. Non c'è uno speciale "statuto AI"; le funzioni AI che soddisfano la definizione del dispositivo sono regolate come SaMD attraverso tre percorsi: 510(k) clearance (dimostrante "equivalenza sostanziale" a un dispositivo predicato), De Novo classificazione (per i nuovi dispositivi a basso rischio senza predicato), e PMA (prevenzione del mercato, per il più alto rischio Classe III). La stragrande maggioranza dei dispositivi AI — circa 97% — entrano via 510(k); solo un paio di dozzine hanno usato De Novo e una manciata PMA (analisi della lista FDA). Il punto di riferimento De Novo è stato IDx-DR nel 2018.
Lo sviluppo recente di definizione è il Predetermined Change Control Plan (PCCP), finalizzato nel dicembre 2024 (FDA). Un PCCP permette a un produttore di pre-specificare e pre-autorizzare una serie di modifiche future del modello - e le parole proprie dell'agenzia catturano il valore: la FDA esamina il PCCP "per garantire la sicurezza e l'efficacia continua del dispositivo senza richiedere ulteriori presentazioni di marketing per l'attuazione di ogni modifica." Nel gennaio 2025 la FDA è andata oltre, emettendo una completa bozza di guida sul ciclo di vita del prodotto totale del software di dispositivo abilitato AI. La postura degli Stati Uniti, in breve: veloce, predicato-driven, imaging-heavy, e ora organizzato intorno alla supervisione del ciclo di vita.
Unione europea — due regimi impilati su un dispositivo
L'UE è il mercato più difficile per l'AI SaMD, perché ora si applicano due regimi regolamentari. In primo luogo, il Medical Device Regulation (MDR). La sua regola di classificazione del software — Rule 11 — è nota. Leggilo direttamente: "I software destinati a fornire informazioni che vengono utilizzate per prendere decisioni con diagnosi o scopi terapeutici sono classificati come classe IIa," escalation a Class III dove una decisione sbagliata potrebbe causare morte o deterioramento irreversibile, o Class IIb per grave deterioramento (MDR, allegato VIII, via EUR-Lex). Sotto le vecchie direttive il software più indipendente auto-certificato come Classe I, senza terzi coinvolti. La regola 11 ha spinto quasi tutto diagnostico e terapeutico SaMD fino a Classe IIa o superiore che forza un Notified Body valutazione della conformità, gestione della qualità ISO 13485 e valutazione clinica. La nostra scansione EUDAMED database trovato AI-keyword dispositivi concentrati in Classe IIa — esattamente l'impronta della regola 11.
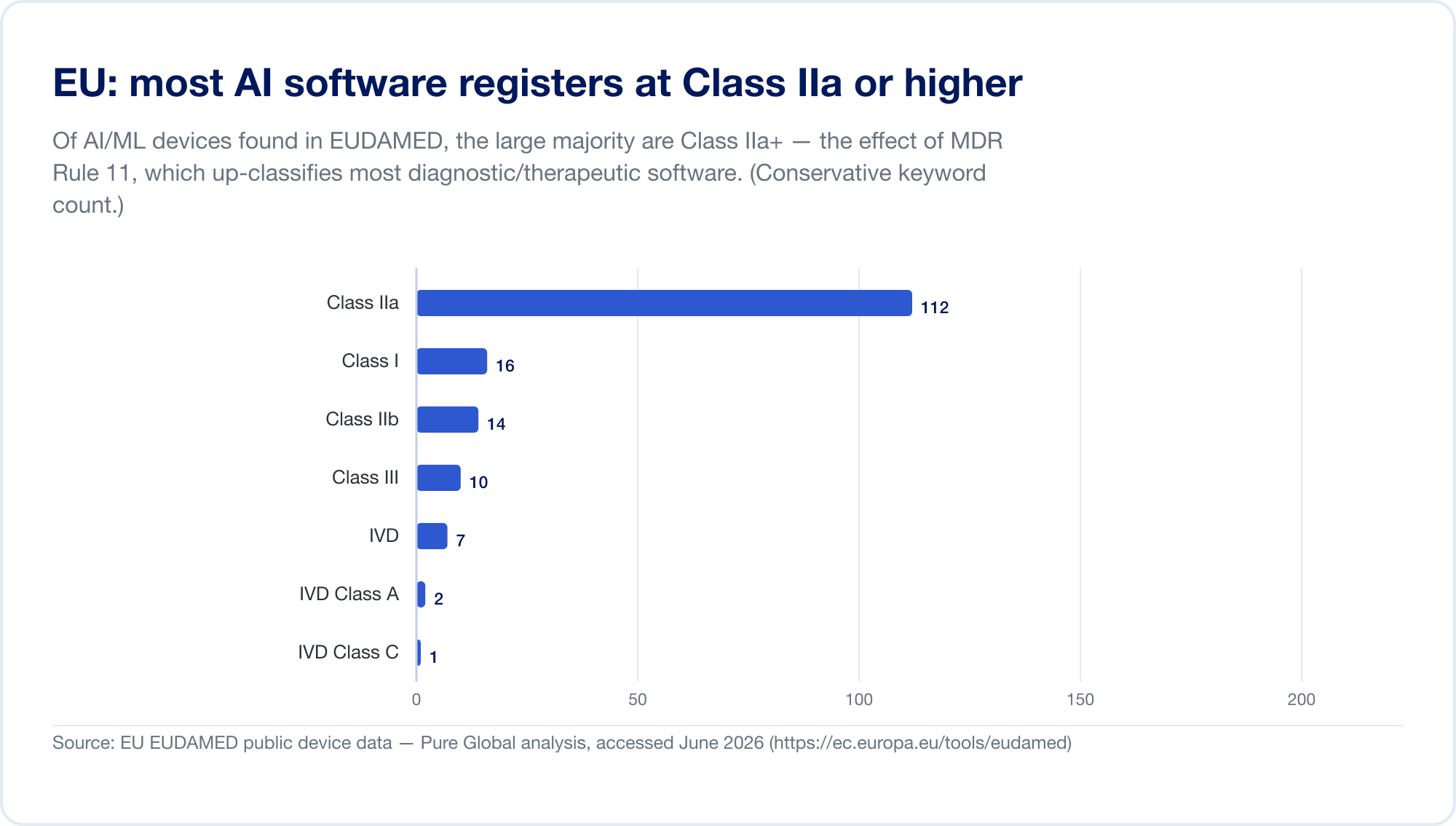
Poi, stratificato sopra, il EU AI Act (regolamento 2024/1689), in vigore dal 1 agosto 2024. Attraverso l'articolo 6, paragrafo 1, un sistema AI è "alto rischio" quando è (o è una componente di sicurezza) un prodotto che richiede già la valutazione della conformità di terzi — che cattura essenzialmente tutta la classe IIa+ Dispositivi medici dell'AI. Fase di obblighi ad alto rischio entro il 2 agosto 2026, con la data per AI incorporata in dispositivi MDR/IVDR fissati a 2 agosto 2027 (una proposta di novembre 2025 "Digital Omnibus" può differire questo al 2028 — trattare le date come in movimento) (artificialentelligenceact.eu). Le penalità arrivano fino a 35 milioni di euro o al 7% del fatturato globale (Articolo 99). Per chiarire la sovrapposizione, il MDCG e il nuovo Consiglio europeo dell'AI hanno pubblicato una FAQ congiunta, MDCG 2025-6, nel giugno 2025 (Commissione europea).
Il vincolo è la capacità. Il numero di MDR Notified Bodies è sceso da circa 80–96 sotto le direttive a circa 50, con solo ~17-19 designati sotto la IVDR; la certificazione MDR ora richiede 13–18 mesi in media, raddoppiando approssimativamente la norma pre-MDR (MedTech Europa). Per AI SaMD, ogni dispositivo compete per scarse Notified Body slot — e dopo 2027/28 avrà bisogno di valutazione di conformità AI Act pure.
Regno Unito — divergente, pragmaticamente
Post-Brexit, l'MHRA ha tracciato un corso volutamente innovativo. Il suo Software e AI come Medical Device Change Programme (roadmap pubblicato il 2022 ottobre) abbraccia undici pacchetti di lavoro, e l'agenzia si è impegnata a consentire PCCP nelle prossime regole di pre-mercato (MHRA). Il suo AI Airlock sandbox regolamentare — il primo del suo genere per i dispositivi medici AI — ha condotto un pilota di quattro progetti nel 2024 e una fase di sette tecnologie da 2 a 2026, con finanziamenti pluriennali ormai impegnati (MHRA). Praticamente, circa il 90% dei dispositivi sul mercato britannico porta ancora il marchio CE, che la Gran Bretagna accetterà fino a 2028–2030; l'MHRA consultata all'inizio del 2026 sul riconoscimento dei marchi CE indefinitamente (MHRA).
Canada — prima a finalizzare le regole ML dedicate
Health Canada è stato tra i primi regolatori con finalized guida pre-mercato dedicata per i dispositivi medicali abilitati al machine-learning - dapprima finalizzata nel 2025 e pubblicata in forma definitiva riveduta in April 2026 - che copre la classe II-IV, adottando i termini chiave IMDRF e introducendo formalmente il PCCP in modo che le modifiche autorizzate non inneschino un nuovo emendamento di licenza (Salute Canada). Il Canada ha coautore dei documenti tri-regolatori fondamentali — GMLP (2021), principi PCCP (2023), e principi di trasparenza (2024) — e ha richiesto la certificazione MDSAP dal 2019.
Australia — riformato in anticipo, riconciliando per AI
L'Australia ha riformato le sue regole software nel febbraio 2021, intagliando le applicazioni per il benessere a basso rischio mentre il software diagnostico up-classifica (i dispositivi attivi per la terapia con una funzione diagnostica spostata nella classe III). La sua consultazione del 2024, Clarificare e rafforzare la regolamentazione dell'AI, ha attirato oltre 600 stakeholder e ha prodotto 14 risultati chiave attualmente in fase di elaborazione (TGA). Il percorso di affidamento di TGA — accettare "comparabili regolatori d'oltremare" — è un grande accelerante, discusso di seguito.
Giappone — costruito per iterazione
Il Giappone regola SaMD come "dispositivi medici programmati" e ha probabilmente il meccanismo più conforme all'intelligenza artificiale di qualsiasi grande mercato: IDATEN, in vigore dal settembre 2020, è la versione giapponese di un protocollo di gestione del cambiamento post-omologazione, che consente ai produttori di modificare le modifiche pre concordate dell'AI (PMDA). Combinato con il DASH per SaMD l'iniziativa e il percorso prioritario SAKIGAKE, il Giappone ha costruito infrastrutture per il software che si evolve - anche se l'assorbimento è modesto: solo 51 SaMD sulla lista del PMDA a partire dal settembre 2025.
Corea del Sud — il movimento più veloce
La Corea e' in piedi. Oltre alle sue autorizzazioni AI 153 nel 2025, ha costruito un quadro giuridico appositamente realizzato: il Digital Medical Products Act (DMPA), efficace gennaio 2025, introduce un meccanismo di cambiamento in stile PCCP e un QMS digitale allineato agli articoli di lavoro IMDRF (Emergo). La Corea ha anche pubblicato la prima linea guida del mondo per i dispositivi medici generativi-AI nel gennaio 2025 e ha approvato il suo primo dispositivo nell'aprile 2026 - e presiede il gruppo di lavoro IMDRF AI/ML. Se vuoi vedere dove si sta dirigendo la regolamentazione globale dell'AI, guarda Seoul.
Singapore — l'hub di affidamento
L'HSA di Singapore supera le sue dimensioni, essendo il regime di reliance più efficiente in Asia. La sua guida software, GL-04 (Revisione 4, Dicembre 2025), copre esplicitamente i dispositivi abilitati alla macchina attraverso il ciclo di vita e richiede una Notifica di Cambiamento quando le prestazioni, gli ingressi di un modello AI o il cambiamento di livello umano-sight (HSA). Crucialmente, HSA riconosce cinque agenzie di riferimento (US FDA, EU Notified Bodies, Health Canada, TGA, Giappone MHLW) e ha stimato che circa il 98% delle applicazioni può utilizzare un percorso abridged; dispositivi con due approvazioni precedenti possono registrarsi attraverso un percorso "immediato" in appena un'ora (Singapore MOH).
Cina — grande, distinto ed esigente
La Cina NMPA tratta seriamente il software di supporto decisionale AI: i suoi principi di revisione CMDE 2021 e il software del catalogo di classificazione 2021-2022 che offre una diagnosi o un trattamento delle unità in Class III, il più alto livello di rischio. La Cina ha approvato il suo primo dispositivo AI classe III nel 2020 e aveva raggiunto circa 154 dispositivi medici AI entro la metà del 2025 (JMIR Informatica medica). In un pacchetto di riforma del 2025 d'ottobre, il NMPA si è impegnato a "semplificare i requisiti di registrazione del cambiamento per i dispositivi medici alimentati dall'IA in cui l'algoritmo di base rimane invariato ma le prestazioni dell'algoritmo sono ottimizzate" — una concessione reale ma stretta rispetto al PCCP statunitense (NMPA). La Cina richiede un agente in paese, un test locale di tipo, e — per molti dispositivi di classe III — dati clinici locali, rendendolo tra i mercati più esigenti in questa relazione.
India, Taiwan e il resto dell'Asia-Pacifico
CDSCO dell'India ha emesso una bozza di guida del software del dispositivo medico nell'ottobre 2025, introducendo un "Protocollo di cambiamento dell'algoritmo" per gli aggiornamenti dell'AI, ma rimane una bozza senza la regola di AI finalizzata ancora (CDSCO Taiwan's TFDA, al contrario, ha una delle suite di guida AI più profonde ovunque — linee guida tecniche CADe/CADx dedicate e guida PCCP-drafting — e dispositivi AI/ML con licenza 166 dal 2020 al 2024. Attraverso l'ASEAN, il modello è dipendenza-plus-localizzazione: Malaysia, Thailandia, Vietnam, Filippine e Indonesia per lo più tratta SaMD come dispositivo generale e appoggiato alle approvazioni di riferimento-paese, con il fast-track del Vietnam insolitamente ampio (accetta anche le approvazioni NMPA e MFDS).
America Latina, Medio Oriente e Africa
Brazil's ANVISA ha importato la logica all'ingrosso dell'UE: la sua Rule 11 sotto RDC 751/2022 rispecchia l'UE, ponendo il software di supporto decisionale in classe II-IV, e RDC 657/2022 è stato il primo della regione SaMD- risoluzione specifica. Un produttore straniero non può contenere una registrazione brasiliana - un locale Brazil Registrazione Holder è obbligatorio e non disponibile (Artixio Il COFEPRIS del Messico ha riabilitato il suo regime di affidamento nel 2025 in un unico Pathway Abbreviato che riconosce tutti i membri dell'IDMRF e del MDSAP, con un obiettivo di 30 giorni lavorativi. La SFDA di Arabia Saudita ha emesso MDS-G010 (novembre 2022) — tra le prime guide mediche dedicate AI/ML ovunque, e citata da alcuni osservatori come il primo enforceable (altri la classificano come non vincolante) — che dirige in modo unico che *"il produttore dovrebbe convalidare localmente i dispositivi medici basati su AI/ML che hanno sviluppato e approvato in altre giurisdizioni"SFDA) — un promemoria che "l'approvazione del paese di riferimento" non è sempre sufficiente. Il UAE ha centralizzato l'approvazione del dispositivo sotto un nuovo Emirates Drug Stabiliment nel gennaio 2025. SAHPRA del Sud Africa ha rilasciato la sua prima comunicazione AI nel settembre 2025 ma non ha ancora iniziato a registrare dispositivi, e l'Agenzia continentale African Medicines — 31 di 55 stati ratificati — non copre ancora dispositivi o AI.
Questo è il cuore del problema: quindici giurisdizioni, quindici risposte. Lo stesso software è Classe II negli Stati Uniti, Classe IIa+ e "alto rischio" nell'UE, Classe III in Cina, Grado 2-3 in Corea, e Classe II-IV in Brasile — ciascuno con i propri requisiti di prova, lingua, socio locale e controllo dei cambiamenti.
Quanto costa e quanto tempo ci vuole
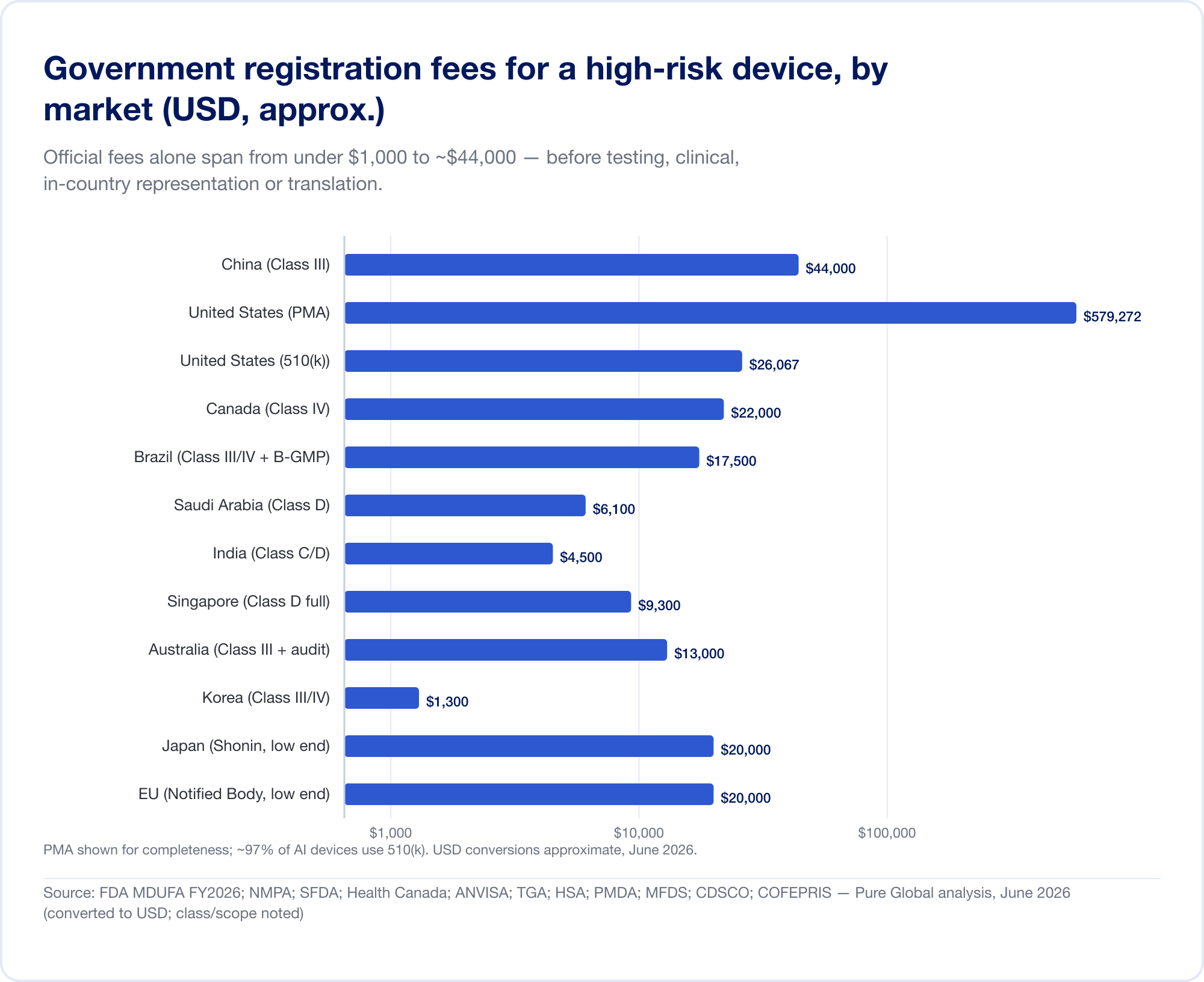
Le differenze di classificazione sopra si traducono direttamente in denaro e mesi. I numeri di titolo riportati di seguito sono tasse di governo e termini realistici per un dispositivo a più alto rischio; escludono i costi sostanziali di test, prove cliniche, traduzione e rappresentazione in paese che spesso nascondono la tassa ufficiale.
La tassa ufficiale è il piccolo numero
Le tasse governative da sole variano da meno di 1.000 USD a circa 44,000 USD per una registrazione ad alto rischio:
- Stati Uniti — Tasse MDUFA FY2026 (verified on FDA.gov): 510(k) $26,067 (piccola impresa $6,517); De Novo $173,782; PMA $579,272; più una tassa di stabilimento annuale $11,423 (FDA).
- Cina — NMPA tasse di registrazione di circa RMB 210,900 (
$30,000) per la classe II e RMB 308,800 ($44,000) per la classe III — le tasse ufficiali più ripide in questa relazione. - Brazil — ANVISA Classe III/IV registrazione di ~BRL 21,000 più una tassa di certificazione B-GMP di BRL 72,804 per i produttori internazionali.
- Canada — Classe III CAD 14,163, Classe IV CAD 30,713 (aprile 2026).
- Arabia Saudita — Tasse SFDA di SAR 15.000–23.000 per classe.
- India — licenza di importazione MD-15 di 3.000 dollari al sito + 1.500 dollari al prodotto per la classe C/D.
- Singapore, Australia, Corea, Giappone — le tasse ufficiali sono relativamente modeste (spesso sotto USD 13.000), ma l'onere di prova e di revisione varia ampiamente.
Il tempo è il numero costoso
Il costo reale è il calendario. I tempi realistici e ad alto rischio si estendono da circa un mese (Canada, Classe III) a 24 mesi nell'UE e quattro a cinque anni in Cina quando è richiesta una sperimentazione clinica locale:
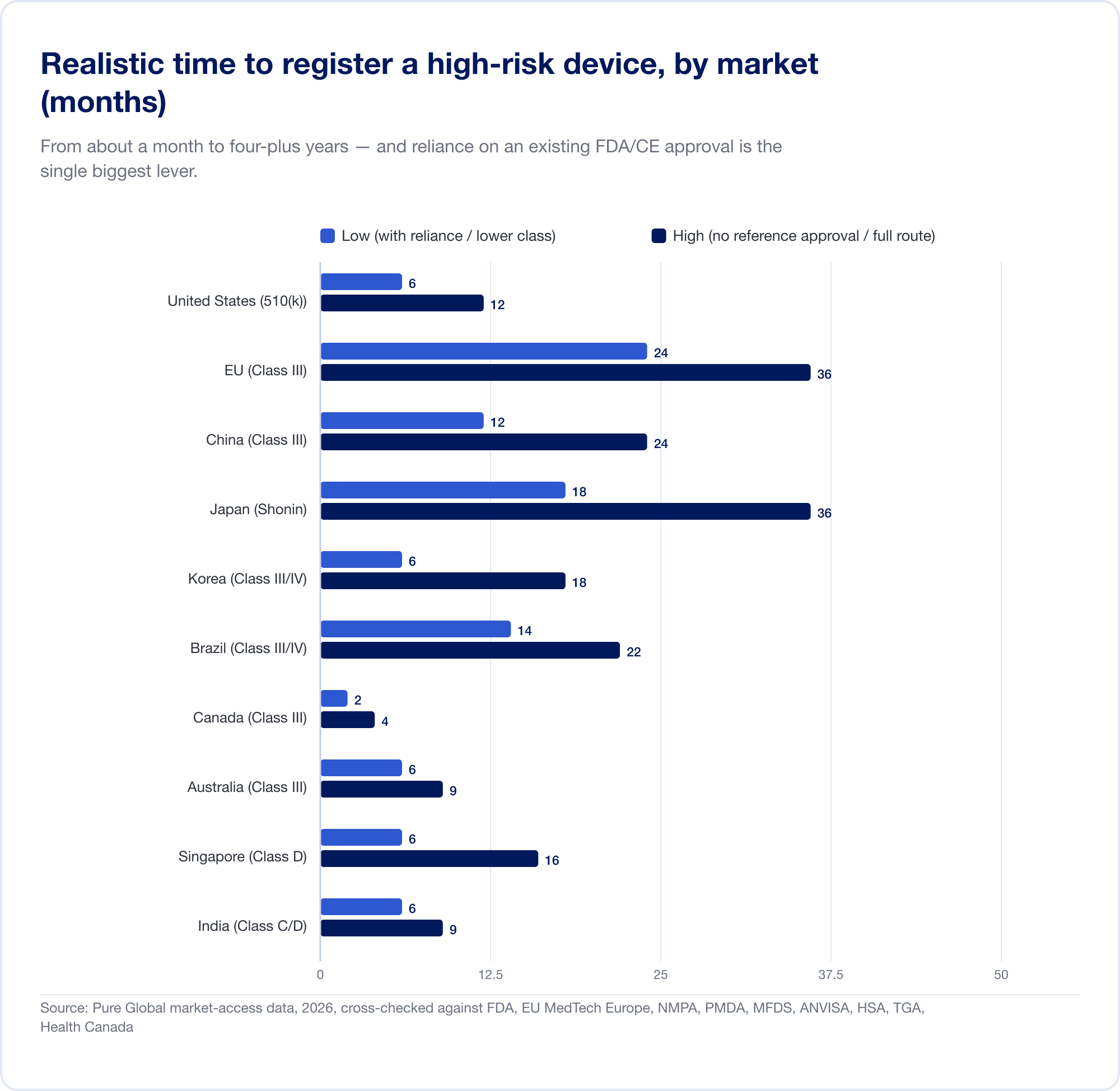
L'UE illustra come la classificazione diventa costosa. Perché la regola 11 converte quello che era un'autodichiarazione di classe I quasi gratuita in una classe IIa+Notified Body impegno, il progetto all-in CE per SaMD comunemente viene eseguito in sei cifre e 13-18 mesi prima di un numero di certificato. Per una startup con una pista misurata in quarti, che non è un elemento di linea — è una minaccia strategica.
La leva: affidamento e torsione specifica AI
Contro queste linee temporali si trova il singolo strumento più potente nell'accesso al mercato globale: reliance. La maggior parte dei mercati al di fuori degli Stati Uniti, l'UE e la Cina si appoggiano su un'approvazione che già avete. I nostri dati di mercato mostrano l'effetto starkly — un alto rischio SaMD che richiede ~8 mesi per registrarsi in Brasile sul percorso standard può scendere verso 6 settimane quando un FDA o un'altra approvazione di riferimento è sfruttato attraverso il percorso di analisi ottimizzata di ANVISA.
Per AI SaMD c'è un tocco specifico e ricorrente che i tavoli dei costi non mostrano. Quando l'algoritmo aggiorna — come la buona AI costantemente fa — la domanda è se tale aggiornamento ha bisogno di una presentazione new. Il PCCP degli Stati Uniti può lasciare la nave di aggiornamenti pre-specificati senza nuova presentazione, risparmiando la tassa $26,067 più la recensione 90–175-day su ogni 510(k evitato), e molto di più per gli integratori PMA evitati. Circa 10% dei dispositivi AI la FDA ha cancellato nel 2025 già incorporato un PCCP. Ma questo risparmio è solo negli Stati Uniti, e questo è il nascondiglio dell'intera relazione.
Quanto costa registrare AI come dispositivo medico in diversi paesi?
Le tasse governative sopra sono solo la metà della fattura. L'altra metà è il lavoro specialistico — costruendo il dossier, tenendo la registrazione locale, mettendo in campo ogni domanda dall'autorità, e archiviando ogni rinnovo e cambiamento dell'algoritmo. Questo è dove l'industria è al suo più opaco: la maggior parte delle consultazioni normative fattura entro l'ora o cita ogni presentazione separatamente, così il vero multi-mercato costo emerge solo dopo l'arrivo degli ordini di cambiamento.
Pure Global è la prima società di accesso al mercato medico-dispositivo a pubblicare una tassa annuale singola e piatta per registrazione. Da USD $2,000 per dispositivo, per mercato, per anno, che una tassa consolida i servizi normalmente fatturati orariamente — in-country rappresentazione, presentazione (su un'approvazione di riferimento), rinnovi, modifiche, e tutta la corrispondenza di autorità sanitaria. Nessun foglio temporale, nessuna sorpresa per e-mail.
Ecco cosa costa avere Pure Global agire come il vostro rappresentante locale e tenere una registrazione AI medico-dispositivo, mercato per mercato. (AI)SaMD di solito atterra nella classe a più alto rischio, quindi la figura superiore si applica nei mercati tiered.)
Pure Global rappresentazione in paese — tassa annuale piatta per dispositivo AI
Un numero trasparente per mercato, per anno — tutto per quella registrazione inclusa.
| Mercato | Ruolo locale Pure Global fornisce | Tassa annuale piatta (USD) |
|---|---|---|
| Stati Uniti | FDA US Agent | $1,000 |
| Unione europea | EUAuthorized Representative | $2,000 |
| Regno Unito | UK Responsible Person (UKRP) | $2,000 |
| Australia | Sponsor TGA | $2,000 |
| Singapore | Registrazione | $2,000 · $3,000 (Classe C/D) |
| Malesia | Authorized Representative | $2,000 · $3,000 (Classe C/D) |
| Thailandia | Authorized Representative | $2,000 · $3,000 (Classe 3/4) |
| Indonesia | Authorized Representative | $2,000 |
| Vietnam | Holder di autorizzazione di mercato | $2,000 |
| Hong Kong | Persona responsabile locale | $2,000 · $3,000 (Classe III/IV) |
| Macau | Porta licenze e registrazione | $2,000 · $3,000 (Classe III) |
| Brasile | Brasile Registrazione titolare (BRH) | $2,000 · $3,000 (Classe III/IV) |
| Messico | Messico Registrazione titolare | $2,000 · $3,000 (Classe II/III) |
| Colombia | Rappresentante INVIMA | $2,000 · $3,000 (Classe IIb/III) |
Flat tassa annuale per la rappresentazione in paese di un AI SaMD; comprende la presentazione su un'approvazione di riferimento, rinnovi, modifiche e corrispondenza dell'autorità. Fonte:Pure Global Master Listino prezzi, 2026 (per registrazione; si applicano sconti multi-registrazione e triennio).
Un tempo sottomissione e compilazione lavoro — dove un mercato ha bisogno di un dossier completo costruito — è pubblicato altrettanto trasparente: una compilazione 510(k) degli Stati Uniti esegue $ 15.000–$20.000, un progetto di documentazione tecnica dell'UE o CER è elencato per classe ($8,000–$30.000), una compilation di registrazione canadese è $3,000–5,000$25,000$ per classe, e a percorso regolatorio Ogni cifra è citata davanti, mai all'ora.
Un esempio di lavoro — un algoritmo di imaging AI, quattro mercati. Prendere un unico strumento di radiologia AI già sgomberato dalla FDA (Classe II) e marcato CE (Classe IIb) che si desidera mantenere in vita attraverso gli Stati Uniti, Unione Europea, Brasile e Singapore per un anno.Pure Global's in-country rappresentazione totale $1,000 (US) + $2,000 (EU) + $3,000 (Brasile) + $3,000 (Singapore) = $9,000 per l'anno — piatto, con ogni rinnovo, modifica e scambio di autorità incluso; si aggiunge un tempo di lavoro di presentazione solo dove un mercato effettivamente richiede un dossier fresco. Questa prevedibilità è il punto: quando le regole differiscono in ogni mercato e il modello continua a cambiare, l'ultima cosa che un produttore ha bisogno è una legge regolamentare che fa anche.
Il paradosso di convergenza
Ecco l'apparente buona notizia. Sotto la patchwork a quindici giurisdizioni si esegue un potente meccanismo di armonizzazione. IMDRF allinea le definizioni e i principi di Good Machine Learning Practice. Il Medical Device Single Audit Program (MDSAP) consente a un unico sistema di controllo di qualità di soddisfare cinque regolatori contemporaneamente — gli Stati Uniti, Canada, Brasile, Giappone e Australia (FDA). E i percorsi di dipendenza si stanno diffondendo velocemente: Singapore accetta cinque agenzie di riferimento e percorsi ~98% delle applicazioni attraverso una revisione ridotta; Brasile, Messico, Australia, Malesia, Vietnam, e il Golfo tutti riconoscono le approvazioni straniere in qualche modo. Nel febbraio 2026 IMDRF pubblicò anche un Reliance Playbook per codificare la pratica (IMDRF N89).
Quali mercati accettano quali approvazioni estere (percorsi di fiducia)
Una licenza FDA o marchio CE è una chiave principale per decine di mercati — ognuno con la propria serratura.
| Mercato | Agenzie di riferimento riconosciute / programmi | Effetto |
|---|---|---|
| Singapore (HSA) | USA FDA, UE NB, Salute Canada, TGA, Giappone MHLW | Abridged/expedited/immediate; ~98% ammissibili |
| Brasile (ANVISA) | TGA, Health Canada, US FDA, Giappone MHLW (Classe III/IV) | "Analisi ottimizzata" ~20-30% più veloce |
| Messico (COFEPRIS) | Tutti i membri IMDRF + partecipanti MDSAP | Cammino aspro, 30 giorni lavorativi |
| Australia (TGA) | USA FDA, Salute Canada, MHLW/PMDA, EU NB, MDSAP | Valutazione della conformità ridotta |
| Malesia (MDA) | USA FDA, Salute Canada, TGA, CE UE, PMDA, HSA, Thai FDA | Percorso di verifica (abridged) + MDSAP |
| Vietnam (MOH) | USA FDA, UE, PMDA, TGA, Salute Canada, MFDS, NMPA | Insolitamente ampio SRA fasttrack |
| Arabia Saudita (SFDA) | FDA / CE solo supportive | Piena recensione tecnico-file ancora richiesta |
| UAE (EDE) | CE, US FDA | Registrazione basata sulla dipendenza |
| MDSAP (un audit) | Stati Uniti, Canada, Brasile, Giappone, Australia | Audit QMS singolo accettato da tutti e cinque |
Fonte: HSA, ANVISA, COFEPRIS, TGA, MDA Malesia, Thai FDA, Vietnam MOH, SFDA, EDE —Pure Global analisi, giugno 2026.
Per un dispositivo static, questo è trasformativo: una forte approvazione — tipicamente FDA o CE — diventa una chiave principale che apre decine di mercati a velocità e costi ridotti. Questo è precisamente la leva di un programma di accesso al mercato ben gestito è costruito per sfruttare.
E qui c'è il paradosso. Per adaptive AI, la chiave principale smette di lavorare esattamente al punto che conta di più — cambiare il controllo. La convergenza è sul dispositivo *; la divergenza è sul AI. Considera lo stesso modello di apprendimento automatico che cerca di aggiornare il suo algoritmo:
- Nel U.S., un PCCP pre-autorizzato permette di spedire l'aggiornamento senza nuova presentazione.
- Nel EU, un cambiamento software "sostanziale" attiva ancora Notified Body re-review — e, stratificato in cima, una valutazione separata di conformità AI Act.
- In Cina, l'aggiornamento è tollerato solo se "l'algoritmo di base rimane invariato"; un vero e proprio riqualificare significa registrazione a pieno cambiamento.
- In Corea, il DMPA consente piani di cambiamento pre-approvati, ma solo nei parametri pre-approvati.
Poiché un'analisi lo mette in modo sfocato, "un PCCP autorizzato dalla FDA non soddisfa gli obblighi dell'UE AI Act, e viceversa" (Berkley Lifesciences). Nessun controllo MDSAP e nessun percorso di affidamento risolve questo problema. Uno sviluppatore AI che vince le autorizzazioni in dieci mercati non ha acquistato la pace; hanno acquistato dieci diversi obblighi di controllo dei cambi, ciascuno innescato ogni volta che il modello migliora. Per una tecnologia la cui proposizione di valore intero è che continua a migliorare, cioè una tassa strutturale e compositiva sul successo — e cade più duro sui piccoli, veloce-moving team che costruiscono i migliori modelli.
La prossima frontiera: modelli generativi di AI e di fondazione
Se l'IA adattativa ha teso il sistema, l'AI generativa minaccia di sopraffarlo. Tutto quanto sopra presuppone un modello addestrato per un uso unico, ben definito previsto — rileva un sanguinamento, misura una frazione di espulsione, contrassegna un nodulo. Modelli linguistici di grandi dimensioni e modelli di fondazione multimodale rompono l'ipotesi in tre modi: sono general-purpose (un modello, molti usi possibili), non-deterministic (lo stesso prompt può dare risposte diverse), e incline a allucinazione (confidente, fluente, sbagliato). Nessuna di queste proprietà si adatta comodamente all'interno di un quadro costruito intorno ad un uso fisso e una versione di riferimento "bloccato".
I regolatori lo sanno. Il WHO ha emesso la prima guida globale dedicata ai grandi modelli multi-modali nel gennaio 2024, avvertendo specificamente su uscite fabbricate, bias di automazione e la difficoltà di validare sistemi formati su dati su Internet (WHO). Il Comitato Digital Health Advisory FDA ha dedicato la sua riunione inaugurale, nel novembre 2024, alle sfide globali del ciclo di vita dei dispositivi generativi (AI-enabled) FDA). E Corea del Sud, caratteristicamente in primo luogo, ha pubblicato la prima linea guida al mondo per i dispositivi medici generativi-AI nel gennaio 2025 e ha autorizzato il suo primo dispositivo nell'aprile 2026.
Ma la guida non è la stessa di un percorso di sdoganamento. A partire dalla metà del 2026, le rotte stabilite — 510(k), De Novo, marcatura CE — assumono ancora un dispositivo che è possibile pin down, prova contro uno standard fisso, e congelare. Un LLM clinico generico non soddisfa nessuno di questi presupposti in modo pulito, motivo per cui la prima ondata di "AI generativo nel settore sanitario" ha raggiunto il mercato principalmente come strumenti amministrativi (scrizioniambient, aiuti di documentazione) che affiancano la definizione del dispositivo, piuttosto che come dispositivi diagnostici chiariti. La frontiera regolamentare per l'IA clinica autonoma e generativa è, in realtà, ancora in fase di elaborazione — e i mercati che lo disegnano prima (Corea oggi; altri da seguire) plasmano come il resto del mondo li copia. Per gli sviluppatori, la lezione pratica è quello di guardare dove le linee sono impostati e progettare la strategia di regolazione, non solo il prodotto, per un obiettivo in movimento.
Il gioco di accesso al mercato
Se il problema è la frammentazione, la risposta è un sistema. Attraverso i modelli sopra, un playbook ripetibile emerge per ottenere un dispositivo medico AI al mondo - e tenerlo lì.
Classifica prima di costruire le prove. Lo stesso prodotto può essere Classe II o Classe III a seconda del mercato e della rivendicazione. Mappa le regole di classificazione dei mercati target first, perché dettano le prove cliniche e tecniche di cui hai bisogno. Sequenziare la domanda e le prove al più rigoroso mercato target evita di ricostruire il dossier in seguito.
Vincere una forte approvazione di ancoraggio - quindi sfruttare deliberatamente la dipendenza. Una licenza FDA o marchio CE vale molto più di un mercato; è la credenziale che sblocca rotte abridged in Singapore, Brasile, Messico, Australia, il Golfo, e oltre. L'arte è conoscere che ancora ogni mercato target riconosce, e routing il dossier di conseguenza. L'Arabia Saudita, che richiede la convalida locale anche dell'AI approvato dall'estero, è il promemoria che la dipendenza è una mappa, non una coperta.
Rappresentanza in paese dove è obbligatorio - che è la maggior parte dei posti. Un produttore straniero non può tenere la propria registrazione in Brasile (BRH), l'UE (Authorized Representative), Cina (agente legale), Giappone (MAH/DMAH), Arabia Saudita, Emirati Arabi Uniti, India e molti altri. Ciascuno richiede un soggetto legale locale di tenere la registrazione e di fronte all'autorità sanitaria. In piedi trenta di tali entità è impraticabile; outsourcing a un singolo partner è come la scala diventa fattibile.
Trattare il controllo del cambiamento come un flusso di lavoro multi-mercato di prima classe. Questa è la disciplina specifica dell'AI. Costruire un PCCP degli Stati Uniti, ma anche mappare come ogni mercato gestisce l'aggiornamento same - e progettare la cadenza di rilascio dell'algoritmo intorno al regime più restrittivo che conta per il vostro business. Il piano del ciclo di vita è ora parte del piano di accesso al mercato.
Eseguire come un'operazione collegata, non trenta filings scollegati. Il costo non è una singola registrazione; è il coordinamento — traduzioni, titolari locali, calendari di rinnovo, notifiche di cambiamento e vigilanza post-mercato attraverso decine di regimi che si allontanano indipendentemente.
Una sequenza tipica nella pratica. Per uno sviluppatore di imaging-AI, il rollout funziona spesso: garantire il FDA 510(k) o CE mark come l'ancoraggio; in parallelo, file nel vostro mercato di casa e un mercato di affidamento rapido (Singapore o Australia) per le entrate bancarie anticipate; utilizzare tali autorizzazioni per aprire le rotte abridged in Brazil, Messico, e il Golfo; poi prendere i dati clinici di alto valore, i tempi alti Nel corso di tutto, un unico piano di controllo del cambiamento viene mantenuto centralmente e mappato alle regole di aggiornamento di ogni mercato, quindi un miglioramento del modello viene archiviato ovunque deve essere, e in nessun luogo non deve essere. L'ordine non è arbitrario; sequenzia il flusso di cassa, il riutilizzo delle prove e i mercati delle scarse risorse deliberatamente.
Questo è il lavoro.Pure Global è costruito per fare: in-country rappresentazione e AI-assisted esecuzione normativa attraverso 30+ mercati, consegnato su una tassa annuale piatta piuttosto che il modello orario aperto a cui l'industria di default. I dati in questa relazione — tratti dalla FDA,EUDAMED, NMPA, PMDA, MFDS, ANVISA e decine di registri nazionali, insieme al nostro mercato-per-mercato costi e timeline dataset — è la stessa intelligenza che usiamo per sequenza di un cliente globale rollout. Il punto di mappare il labirinto questo accuratamente è di essere in grado di camminare un cliente attraverso di esso rapidamente.
Conclusione: quattro cose da togliere
AI è ora una categoria di dispositivi mainstream, e i dati lo dimostrano. Da circa 1 in 700 autorizzazioni FDA nel 2019 a 1 in 28 nel 2025, con 1.500-plus dispositivi AI autorizzati negli Stati Uniti da soli e programmi nazionali in Corea, Cina, Taiwan e Giappone scaling veloce, AI SaMD ha attraversato dalla novità alla norma.
Ogni regolatore lo riclassifica, e la maggior parte sono convergenti sulla supervisione del ciclo di vita. Regola 11 nell'UE e in Brasile, classe III in Cina, alto rischio sotto la legge UE sull'AI — l'IA diagnostica e terapeutica è trattata come seria, e la risposta condivisa è il controllo totale del ciclo di vita del prodotto, GMLP e piani di cambiamento prestabiliti.
La fiducia fa un viaggio di approvazione, tranne per la parte AI. Il macchinario di armonizzazione (IMDRF, MDSAP, linee di affidabilità) permette di sbloccare in modo autentico decine di mercati. Ma l'adaptive-AI cambia controllo si diverte bruscamente, quindi un'unica approvazione non rimane valida ovunque mentre il modello si evolve. Questo divario è la sfida operativa di definizione del campo.
Il bordo competitivo è la macchina di registrazione, non la registrazione. Quando le regole differiscono in ogni mercato e cambiano ogni anno, il vantaggio durevole appartiene a coloro che possono registrare velocemente, ovunque, e mantenere ogni approvazione viva attraverso ogni aggiornamento del modello — come un sistema collegato.
Parla con noi
Se state costruendo o scalando un dispositivo medico AI e pesando quali mercati entrare, in che ordine, e come mantenere ogni approvazione valida come il vostro modello migliora, che è esattamente il problema che risolviamo.# Talk to Pure Global circa un piano di accesso al mercato costruito sui dati sopra — o esplorare il nostro guide di registrazione di mercato-per-mercato per andare più a fondo su qualsiasi singolo paese.
Fonti
Organi di governo e di armonizzazione citati in linea sopra; riferimenti chiave raggruppati di seguito. Tutte le cifre sono datate; le cifre sulle dimensioni del mercato e sulle previsioni sono stime di terzi la cui portata varia e deve essere letta come range.
Definizioni, quadri e principi guida
- IMDRF — SaMD: Definizioni chiave (N10, 2013); * Categorizzazione del rischio* (N12, 2014); * Dispositivi medici abilitati a LML: Termini chiave* (N67, 2022); * Principi di guida GMLP* (N88, 2025); Reliance Playbook (N89, 2026). imdrf.org
- US FDA — Software come dispositivo medico (SaMD); * Quadro regolamentare proposto per le modifiche alla base di AI/ML SaMD* (2019); AI/ML Action Plan (2021). fda.gov
- QUI — Etica e governance dell'AI per la salute (2021); * Considerazioni regolamentari sull'AI per la salute* (2023); LMM guida (2024); * Buone pratiche di sollievo*, TRS 1033 allegato 10 (2021). chi.int
Stati Uniti *
- FDA — Artificial Intelligence-Enabled Medical Device s lista (aggiornamento Q1-2026, 1.524 dispositivi; ~ 76% radiologia); PCCP orientamento finale (dice 2024); Digital Health Advisory Committee on Genetive AI (nov 2024); MDUFA FY2026 commissioni. fda.gov · The Imaging Wire (analisi radiologia-share, 2026)
Unione europea e Regno Unito
- EUR-Lex — Regolamento (UE) 2017/745 (MDR), Allegato VIII Regola 11; Regolamento (UE) 2024/1689 (AI Act), Articoli 6, 99, 113. Commissione europea — MDCG 2019-11 Rev.1 & MDCG 2025-6.MedTech Europa & Team-NB (Notified Body capacità). MHRA — Software & AI Change Programme; AI Airlock; guida di riconoscimento CE. Gov.uk
Canada, Australia, Giappone, Corea
- Salute Canada — Guida pre-mercato per dispositivi medici abilitati a ML (Apr 2026). canada.ca
- TGA — riforme software (2021); risultati di consultazione dell'AI (2024–26). tga.gov.au
- PMDA/MHLW —SaMD guida; DASH per SaMD; IDATEN. pmda.go.jp
- MFDS — 2025 Rapporto di approvazione (153 dispositivi AI); DMPA; orientamento generativo-AI. mfds.go.kr; bioin.or.kr
Asia-Pacifico, LatAm, MEA
- HSA Singapore — GL-04-R4; agenzie di affidamento/riferimento. hsa.gov.sg; moh.gov.sg
- NMPA Cina — AI classificazione & Oct 2025 riforma. nmpa.gov.cn · JMIR Medical Informatics (154 AIMDs, 2026).
- CDSCO India — Guida del software MD (Oct 2025). TFDA Taiwan — Guida CADe/CADx; J. Formos. Med. Assoc. (166 licenze).
- ANVISA Brasile — RDC 751/2022 (Regola 11), RDC 657/2022, IN 290/2024. gov.br/anvisa
- COFEPRIS Messico — Pathway abbreviato (2025). SFDA Arabia Saudita — MDS-G010 (2022). UAE EDE — Decreto federale 38/2024. SAHPRA Sud Africa — Comunicazione AI (2025).
Market, aziende, brevetti, sicurezza
- Dimensione del mercato: Grand View Research; MarketsandMarkets; The Business Research Company; Mordor Intelligence; Fortune Business Insights (2025–26).
- Finanziamenti e aziende: Rock Health (2025 finanziamenti digital-health); CB Insights; società FY2025 filings (Tempus AI, iRhythm, HeartFlow, Butterfly Network, Lunit, VUNO); Aidoc (Series E).
- Brevetti: WIPO Technology Trends (2019) & Generative AI Patent Landscape (2024); CNIPA.
- Sicurezza: NEJM (pulse-oximeter bias, 2020); JAMA Internal Medicine (Epic Sepsis Model, 2021); JAMA Health Forum & JAMA Network Open (Ricordi AI, 2025); FDA impulso-ossimetro guida.
- Commercio: OCSE-IMF-WTO Measuring Digital Trade (2021); OMC; UNCTAD.
- Pure Global costi di accesso di mercato proprietario & dataset timeline (2026); open FDA,EUDAMED Analisi di database MFDS —Pure Global, giugno 2026.



Parliamo,
Ovunque tu sia.
Se siete alla ricerca di maggiori informazioni o pronti a collaborare con noi, siamo qui per guidarvi attraverso ogni fase del processo normativo.
Contattaci