
AI als Medizinprodukt: Die globale Karte der Regulierung, Registrierung und des Marktzugangs
Künstliche Intelligenz ist die am schnellsten wachsende Klasse medizinischer Geräte in der Geschichte – und die am stärksten divergierende Regulierung. Das US-amerikanische FDA listet jetzt 1.524 AI-fähige Geräte auf; Südkorea genehmigte 153 in einem einzigen Jahr. Dennoch gehört dieselbe Software zur Klasse II in den USA, zur Klasse IIa+ und „hohes Risiko“ im EU und zur Klasse III in China – jeweils mit eigenen Nachweis-, lokalen Inhaber- und Änderungskontrollregeln. Dieser Bericht stellt dar, wie jede große Regulierungsbehörde AI als medizinisches Gerät klassifiziert, genehmigt und überwacht, mit Registrierungskosten, Zeitplänen und Vertrauenswegen in über 30 Märkten und dem Leitfaden, um diese zu erreichen, ohne jedes Mal das Dossier neu erstellen zu müssen.
Ein evidenzbasierter Praxisleitfaden, der zeigt, wie Regulierungsbehörden weltweit künstliche Intelligenz im Gesundheitswesen klassifizieren, genehmigen und überwachen – und wie man mehr als 30 Märkte erreicht, ohne das Dossier jedes Mal neu erstellen zu müssen.
TL;DR
Künstliche Intelligenz ist zur am schnellsten wachsenden Kategorie medizinischer Geräte in der Geschichte geworden. Die öffentliche Liste der AI-fähigen Geräte der US-amerikanischen Food and Drug Administration erreichte bis Mitte 2026 1.524 Zulassungen, davon etwa drei Viertel in der Radiologie (FDA). Nach unserer eigenen Analyse der FDA 510(k)-Datenbank stieg der Anteil von AI an allen Freigaben von ca. 1 von 700 im Jahr 2019 auf etwa 1 von 28 im Jahr 2025. Südkorea allein hat 153 AI-Geräte im Jahr 2025 zugelassen (MFDS).
Aber dieselbe Software, die durch eine Agentur läuft, kann bei der nächsten ins Stocken geraten. Hier ist die Argumentation dieses Berichts in fünf Punkten:
- AI ist ein Gerät und das meiste davon ist „Software als medizinisches Gerät“ (SaMD). Wenn Software eine Diagnose stellt, eine Triage durchführt oder eine Behandlung empfiehlt, wird sie wie ein Skalpell oder ein Scanner reguliert – in Dutzenden von Gerichtsbarkeiten, jede mit ihren eigenen Regeln.
- Die ganze Welt stuft es hoch. Die MDR-Regel 11 von EU, Brasiliens Regel 11, Chinas Klasse-III-Standard für diagnostische AI – Diagnose- und Therapiesoftware fällt fast überall in eine Klasse mit höherem Risiko, die eine Überprüfung durch Dritte erfordert.
- Das adaptive AI hat das Modell der einmaligen Genehmigung durchbrochen. Ein Modell, das nach der Auslieferung weiterhin Änderungen lernt. Deshalb haben die Regulierungsbehörden neue Mechanismen erfunden – den US-amerikanischen Predetermined Change Control Plan, Japans IDATEN, Koreas DMPA –, die nicht zueinander passen.
- Reliance konvergiert, AI divergiert. Ein Netz von Vertrauens- und Anerkennungspfaden soll es ermöglichen, mit einer Zulassung viele Märkte zu erschließen. Es funktioniert – für statische Geräte. Für adaptives AI gilt: „Ein vom FDA autorisiertes PCCP erfüllt nicht die Verpflichtungen des EU AI Act und umgekehrt“ (Berkley Lifesciences).
- Die Gewinner industrialisieren also die Multi-Markt-Registrierung. Der Wettbewerbsvorteil liegt nicht mehr in einer einzelnen Freigabe; Es ist die operative Maschine, die aus einer Genehmigung dreißig macht und jede einzelne gültig hält, während sich der Algorithmus weiterentwickelt.
Dies ist die Lücke, die Pure Global schließt – Vertretung im Land und AI-unterstützte regulatorische Umsetzung in über 30 Märkten zu einer jährlichen Pauschalgebühr. Der Rest dieses Berichts ist die Karte.
Was „AI als Medizinprodukt“ eigentlich ist
Beginnen Sie mit dem Wort, das die schwere Arbeit übernimmt: Gerät. Wenn eine Software „zur Verwendung bei der Diagnose, Behandlung oder Vorbeugung von Krankheiten bestimmt ist“, handelt es sich in fast allen Rechtssystemen der Welt um ein medizinisches Gerät – unabhängig davon, ob sie auf einem Chip in einem MRT-Scanner oder als App im Browser eines Radiologen läuft. AI hat in den meisten Ländern kein eigenes Statut; es übernimmt den gesamten Apparat des Medizinprodukterechts.
Die Ankerdefinition stammt vom Internationalen Forum der Regulierungsbehörden für Medizinprodukte (IMDRF), dem Gremium, in dem die großen Agenturen ihren Wortschatz harmonisieren. In seinem grundlegenden Dokument aus dem Jahr 2013 definierte IMDRF Software als Medizinprodukt (SaMD) als „Software, die für einen oder mehrere medizinische Zwecke bestimmt ist und diese Zwecke erfüllt, ohne Teil eines medizinischen Hardwaregeräts zu sein“ (IMDRF N10). Das FDA hat diese Sprache wörtlich übernommen und zieht eine dreifache Unterscheidung, die für alles nachgelagerte von Bedeutung ist (FDA):
- SaMD – Software, die ist das medizinische Gerät (ein Thorax-Röntgen-Triage-Algorithmus, ein Diabetiker-Retinopathie-Detektor). Hier leben die meisten klinischen AI.
- SiMD — Software in einem medizinischen Gerät, integraler Bestandteil der Hardware (die Firmware, die eine Infusionspumpe betreibt).
- Software gewohnt herstellen oder warten ein Gerät, das wiederum anders geregelt wird.
Speziell für AI definiert das Schlüsselbegriffsdokument 2022 von IMDRF ein durch maschinelles Lernen gestütztes medizinisches Gerät als „ein medizinisches Gerät, das maschinelles Lernen teilweise oder vollständig nutzt, um seinen beabsichtigten medizinischen Zweck zu erreichen“ (IMDRF N67).
Die Unterscheidung, die alles kaputt macht: gesperrt vs. adaptiv
Die traditionelle Geräteregulierung basiert auf einem einfachen Geschäft: Sie beweisen einmal – für ein festes Design –, dass ein Gerät sicher und effektiv ist, und dieses Design bleibt dann bestehen. AI widerlegt diese Annahme. Das entscheidende Diskussionspapier des FDA aus dem Jahr 2019 hat die Grenze genau gezogen. Ein „gesperrter“ Algorithmus ist einer, der „liefert jedes Mal das gleiche Ergebnis, wenn die gleiche Eingabe darauf angewendet wird, und ändert sich bei der Verwendung nicht.“ – eine Nachschlagetabelle, ein Entscheidungsbaum, ein eingefrorener Klassifikator (FDA, 2019). Ein adaptiver oder kontinuierlich lernender Algorithmus ändert sich nach seiner Bereitstellung.
Diese einzige Eigenschaft – Software, die sich in diesem Bereich selbst verbessert – ist der Grund, warum AI ein Jahrzehnt neuer Regulierung benötigte. Wenn das Produkt, das Sie im Januar genehmigt haben, nicht das Produkt ist, das im Juni in einem Krankenhaus verwendet wurde, was genau haben Sie dann genehmigt? Jeder Rahmen in diesem Bericht ist im Grunde ein Versuch, diese Frage zu beantworten, und die gemeinsame Antwort ist der gesamte Produktlebenszyklus (TPLC): die gesamte Lebensdauer des Geräts zu überwachen, nicht nur den Moment der Freigabe.
Wie die Regulierungsbehörden entscheiden, wie genau sie suchen
Der Risikokategorisierungsrahmen von IMDRF aus dem Jahr 2014 legte die Logik fest, der die Welt jetzt folgt: Die Prüfung, die ein SaMD verdient, hängt von zwei Faktoren ab: die Bedeutung der bereitgestellten Informationen (macht es informieren, fahren, oder diagnostizieren/behandeln?) und die Schwere der Gesundheitssituation (nicht ernst, ernst oder kritisch) (IMDRF N12). Eine App, die Dehnübungen gegen Rückenschmerzen vorschlägt, und ein Algorithmus, der eine Gehirnblutung meldet, sind nicht dasselbe regulatorische Tier, und dieses zweiachsige Raster ist der Grund dafür.
Der Weltgesundheitsorganisation fügte das ethische Gerüst hinzu. Sein Bericht 2021 Ethik und Governance künstlicher Intelligenz für die Gesundheit sechs Prinzipien festlegen – Autonomie schützen; Wohlbefinden und Sicherheit fördern; Gewährleistung von Transparenz und Erklärbarkeit; Verantwortung und Rechenschaftspflicht fördern; Gewährleistung von Inklusivität und Gerechtigkeit; und reaktionsfähiges, nachhaltiges AI fördern (WER). Die WHO folgte im Jahr 2023 mit regulatorischen Überlegungen und veröffentlichte im Januar 2024 die ersten globalen Leitlinien, die direkt auf generative AI und große multimodale Modelle abzielten (WER).
Wie wir hierher gekommen sind
Die medizinischen Geräte von AI brachten keinen einzigen Durchbruch; Sie nahmen über einen Zeitraum von etwa einem Jahrzehnt von Land zu Land zu. Die folgenden Meilensteine zeigen, wie aus einer Definition im Jahr 2013 dedizierte Lebenszyklusregeln auf fünf Kontinenten wurden.
Eine globale Zeitleiste der AI-Regulierung für Medizinprodukte (2013–2026)
In kaum einem Jahrzehnt entwickelte sich AI SaMD von einer IMDRF-Definition zu dedizierten Lebenszyklusregeln auf fünf Kontinenten.
| Datum | Meilenstein |
|---|---|
| Dezember 2013 | IMDRF N10 definiert „Software als medizinisches Gerät“ (SaMD) |
| Januar 2017 | FDA löscht Arterys – erste Cloud + klinisches Deep-Learning-Tool (510(k)) |
| April 2018 | FDA autorisiert IDx-DR – erste autonome AI-Diagnose (De Novo) |
| Mai 2018 | Korea MFDS genehmigt VUNO Med-BoneAge – Koreas erstes AI-Gerät |
| Dezember 2018 | Japan PMDA genehmigt EndoBRAIN (Klasse III) – Japans erstes AI SaMD |
| April 2019 | FDA-Diskussionspapier zu Änderungen an AI/ML SaMD (gesperrt vs. adaptiv) |
| 2020 | China NMPA genehmigt DeepVessel FFR – erstes AI-Gerät der Klasse III; Japan führt IDATEN + DASH für SaMD ein |
| Januar 2021 | FDA AI/ML Aktionsplan; Juni 2021 Sechs ethische Grundsätze der WHO |
| Okt. 2021 | GMLP – 10 Leitprinzipien (FDA + Health Canada + UK MHRA) |
| 2022 | Saudi SFDA MDS-G010 – frühe dedizierte AI-Geräterichtlinie (von einigen als die erste „durchsetzbare“ bezeichnet); IMDRF N67 ML-Begriffe; Brasilien RDC 751/657 |
| August 2024 | EU AI-Gesetz tritt in Kraft; WHO-LMM-Leitfaden (generative-AI) (Januar 2024) |
| Dez. 2024 | FDA stellt die PCCP-Leitlinien (Predetermined Change Control Plan) fertig |
| Januar 2025 | IMDRF N88 GMLP final; Korea gibt die weltweit erste generative AI-Geräterichtlinie heraus; FDA AI Lebenszyklus-Entwurfsleitfaden |
| Februar 2026 | IMDRF N89 Reliance Playbook |
| April 2026 | Health Canada stellt Leitlinien für ML-Geräte fertig; Korea genehmigt das erste generative AI-Gerät |
Quelle: Zusammengestellt aus IMDRF, US, FDA, EU, NMPA, PMDA, MFDS, SFDA, ANVISA, Health Canada und WHO-Primärdokumenten – Pure Global, Juni 2026.
Einige Momente verdienen Hervorhebung. Im Januar 2017 hat das FDA Arterys freigegeben – das erste klinische Tool, das Cloud Computing und Deep Learning kombiniert (PR-Newswire). Dann, im April 2018, kam der Wendepunkt: Das FDA autorisierte IDx-DR, das erste AI, das eine Diagnose autonom liefern darf, ohne dass ein Arzt das Ergebnis interpretiert – ein Diabetiker-Retinopathie-Screening für die Primärversorgung, das in seiner entscheidenden Studie eine Sensitivität von 87,2 % und eine Spezifität von 90,7 % erreichte (npj Digitale Medizin). Innerhalb weniger Monate genehmigten Korea (VUNO Med-BoneAge, Mai 2018) und Japan (EndoBRAIN, Dezember 2018) ihre eigenen ersten Produkte; China folgte im Jahr 2020 mit DeepVessel FFR, seinem ersten AI-Gerät der Klasse III (npj Digitale Medizin).
Das regulatorische Gerüst wurde zwischen 2021 und 2025 aufgeholt: die trilateralen Prinzipien der Guten Praxis des maschinellen Lernens im Oktober 2021, der AI Act der EU, der im August 2024 in Kraft trat, die endgültige FDA-Leitlinie zum vorab festgelegten Änderungskontrollplan im Dezember 2024 und – um Ihnen zu zeigen, wie schnell sich die Grenze verschiebt – Koreas weltweit erste Richtlinie für generative AI-Geräte im Januar 2025 (BioWorld). Bis Februar 2026 hatte IMDRF ein globales Reliance Playbook veröffentlicht, um den Regulierungsbehörden dabei zu helfen, sich gegenseitig auf ihre Arbeit zu stützen (IMDRF N89).
Die Daten: wie groß, wie schnell, wo
Die Clearance-Kurve biegt stark nach oben ab
Der beste Maßstab für die Durchdringung von AI in der Medizin ist die öffentliche FDA-Liste „Künstliche Intelligenz-fähige medizinische Geräte“, die mit ihrer neuesten Aktualisierung (die Daten bis zum ersten Quartal 2026 widerspiegelt) 1.524 Zulassungen erreichte (FDA). Um die Geschwindigkeit zu sehen, haben wir die zugrunde liegende FDA 510(k)-Datenbank direkt analysiert. Die identifizierten AI/ML-Freigaben stiegen von einer Handvoll Ende der 2010er Jahre auf 115 im Jahr 2025 – und, noch bezeichnender, der Anteil von AI stieg von 0,14 % aller 510(k)-Freigaben im Jahr 2019 auf 3,57 % im Jahr 2025, ein etwa 25-facher Anstieg des Anteils in sechs Jahren.
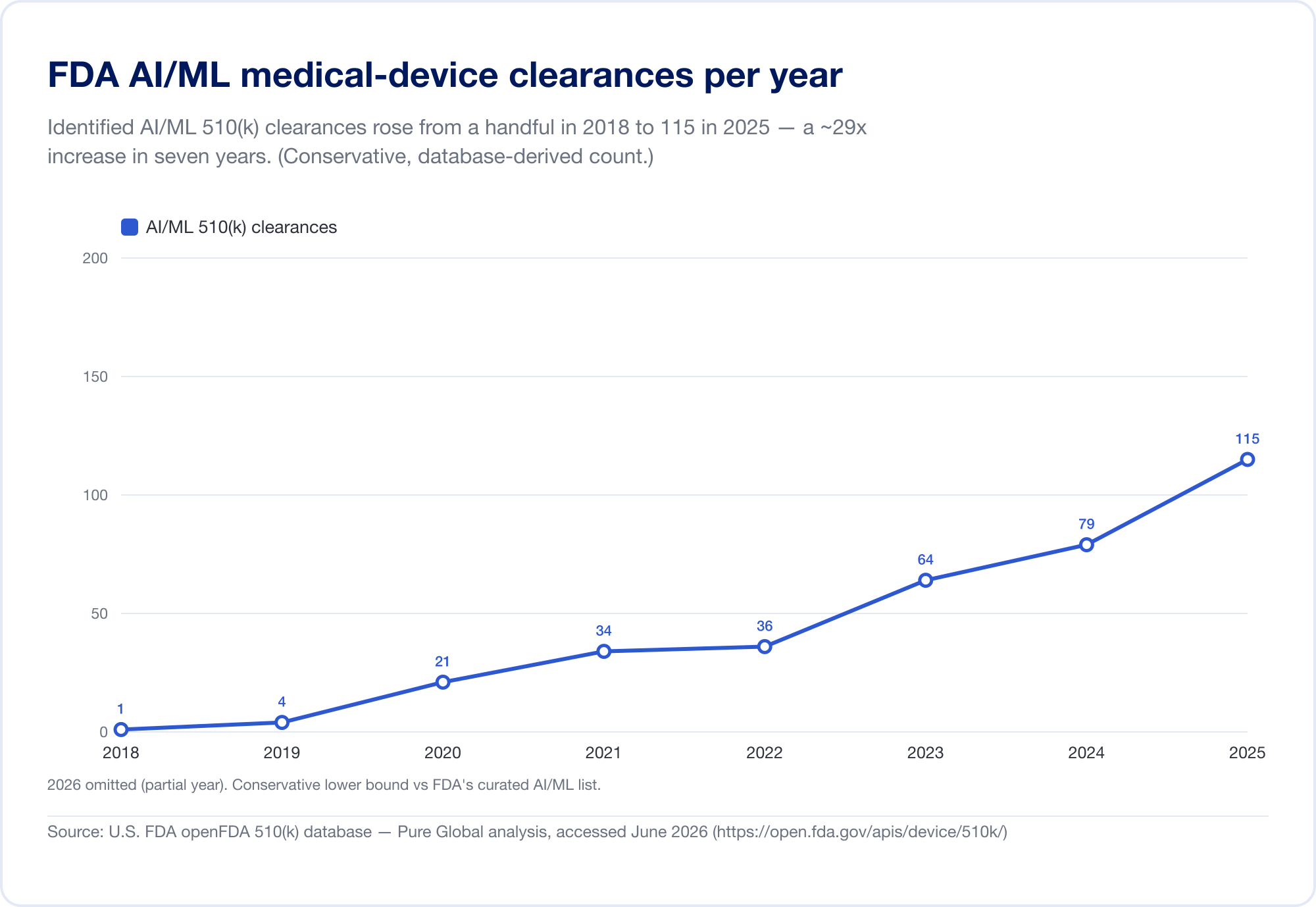
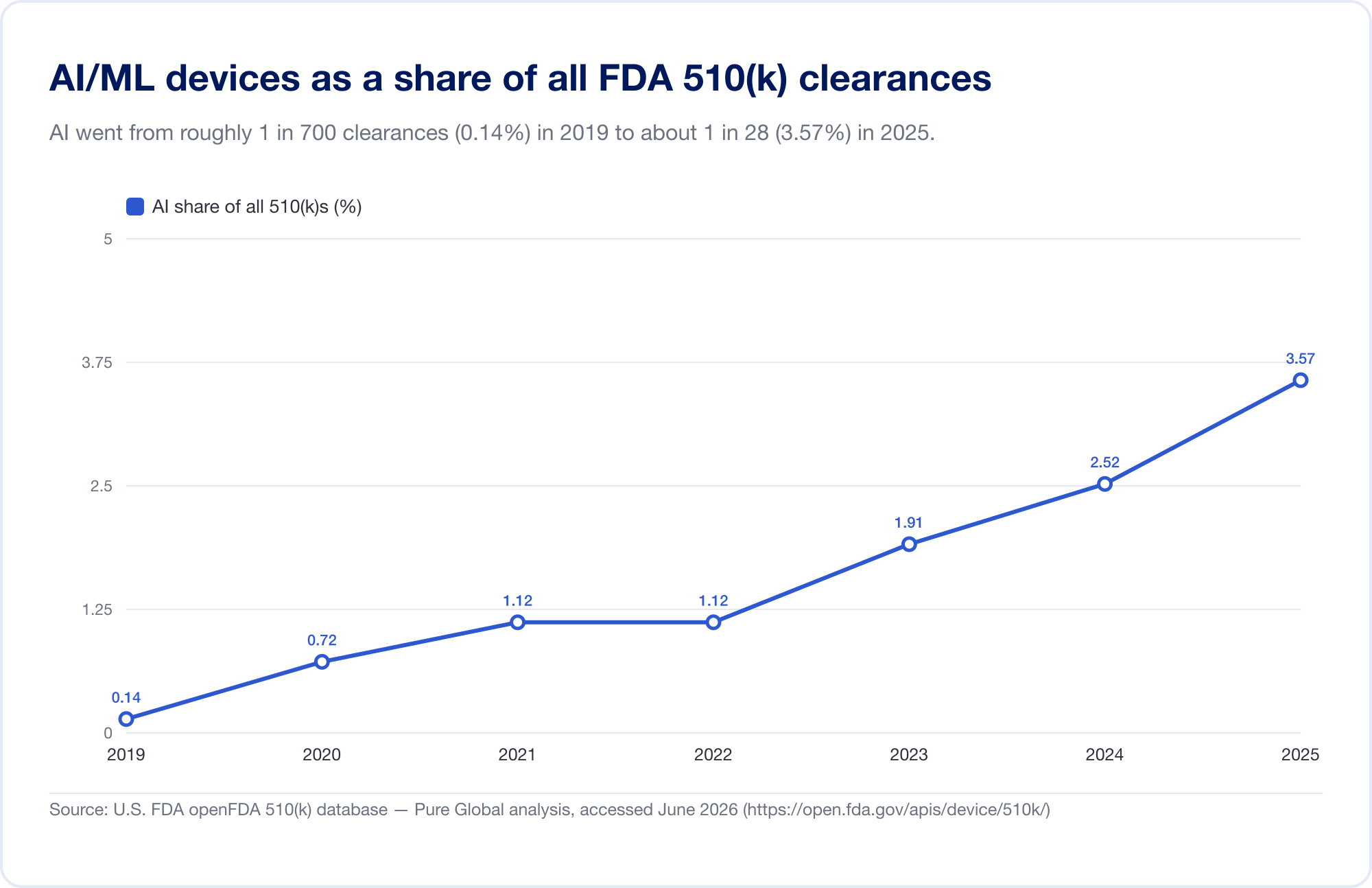
(Unsere aus der Datenbank abgeleitete Zählung ist bewusst konservativ – enger als die kuratierte Liste des FDA, da viele AI-Radiologie-Tools unter Produktcodes stehen, deren Text nie „AI“ sagt. Wir verwenden ihn für Trend und Geografie; die eigene Liste des FDA ist die Gesamtüberschrift. Quelle: openFDA 510(k)-Datenbank – Pure Global-Analyse, Juni 2026.)
Es ist überwiegend eine Bildgeschichte – vorerst
AI in der Medizin heißt heute meist AI in der Radiologie. Auf der Liste des FDA entfallen auf die Radiologie ca. 76 % aller AI-Zulassungen (Der Bildgebungsdraht); In unserer eigenen Clearance-Stichprobe ist die Konzentration sogar noch höher, wobei Herz-Kreislauf und Gastroenterologie (Endoskopie) mit Abstand an zweiter und dritter Stelle liegen. Der Grund ist struktureller Natur: Die Bildgebung ist digital, reichlich vorhanden und gekennzeichnet, und der 510(k)-Pfad ermöglicht es einem neuen Algorithmus, einen vorhandenen als Prädikat zu zitieren. Pathologie, kardiologische Signale und klinische Textmodelle nehmen zu, aber der Schwerpunkt bleibt das Bild.
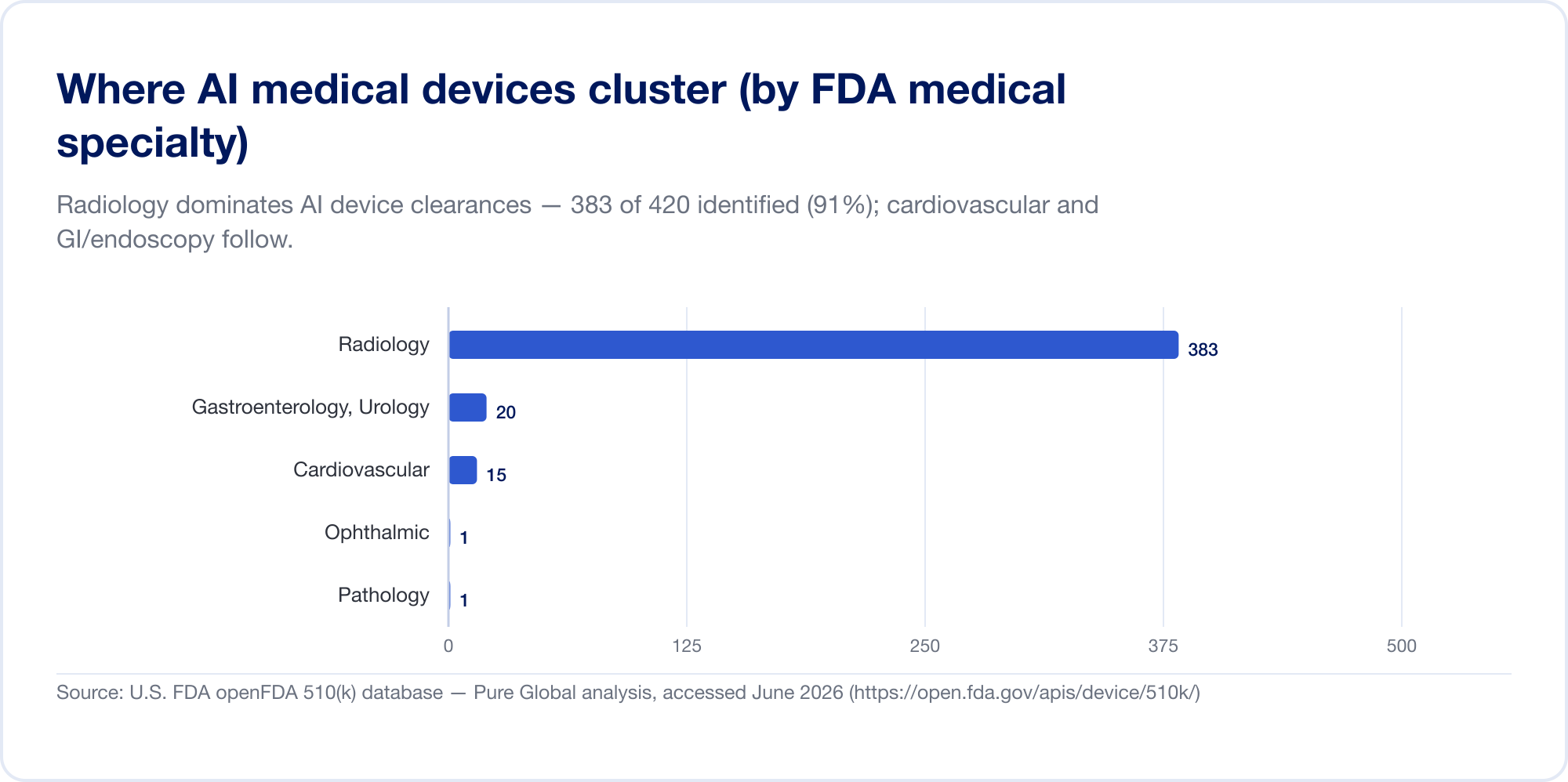
Die Innovatoren sind überall; Die Märkte gibt es überall sonst
Hier ist die Tatsache, die jeden Kommerzialisierungsplan neu gestalten sollte. AI-Medizinprodukte, die in den USA zugelassen sind, stammen aus mindestens 16 Ländern. In unserer Analyse der AI 510(k)-Antragsteller liegen die Vereinigten Staaten an der Spitze, aber Südkorea, Israel, Kanada, Frankreich, China, Deutschland, das Vereinigte Königreich, die Niederlande, Taiwan und Japan liefern jeweils aussagekräftige Zahlen.
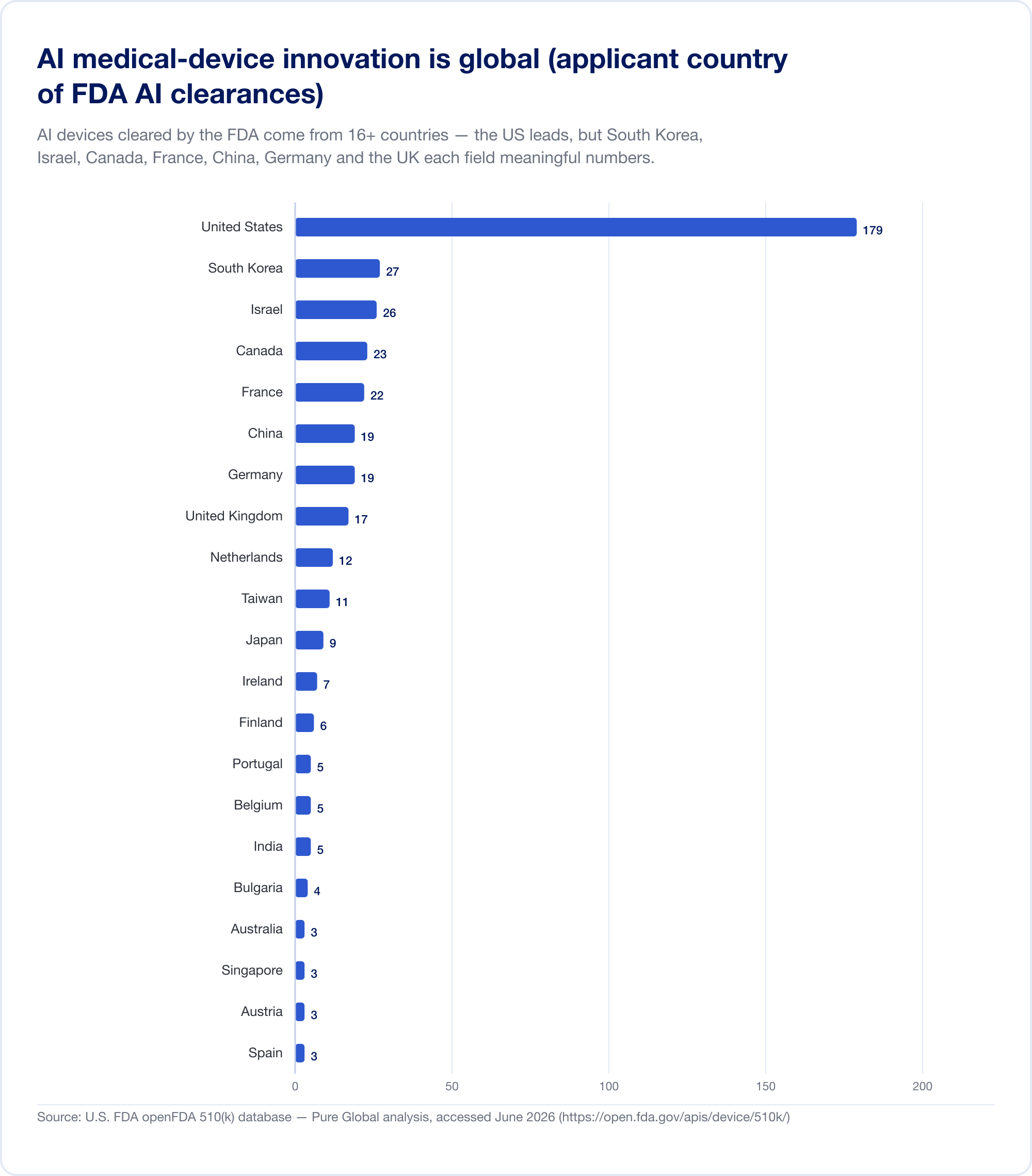
Die nationale Produktion bestätigt das Muster. Südkorea autorisierte (Genehmigungen und Zertifizierungen zusammen) 64 AI-Geräte im Jahr 2023, 108 im Jahr 2024 und 153 im Jahr 2025 — ein Anstieg um 41,6 %, wobei 77,7 % im Inland hergestellt wurden (MFDS). China hatte bis Mitte 2025 grob 154 AI-Medizinprodukte zugelassen, etwa 80 % davon in der höchsten Risikoklasse III (JMIR Medizinische Informatik). Taiwan lizenzierte 166 AI/ML-Geräte zwischen 2020 und 2024 (J. Formos. Med. Assoc.). Japan hatte im September 2025 51 AI-basierte SaMD auf der PMDA-Liste (Globale Gesundheit und Medizin).
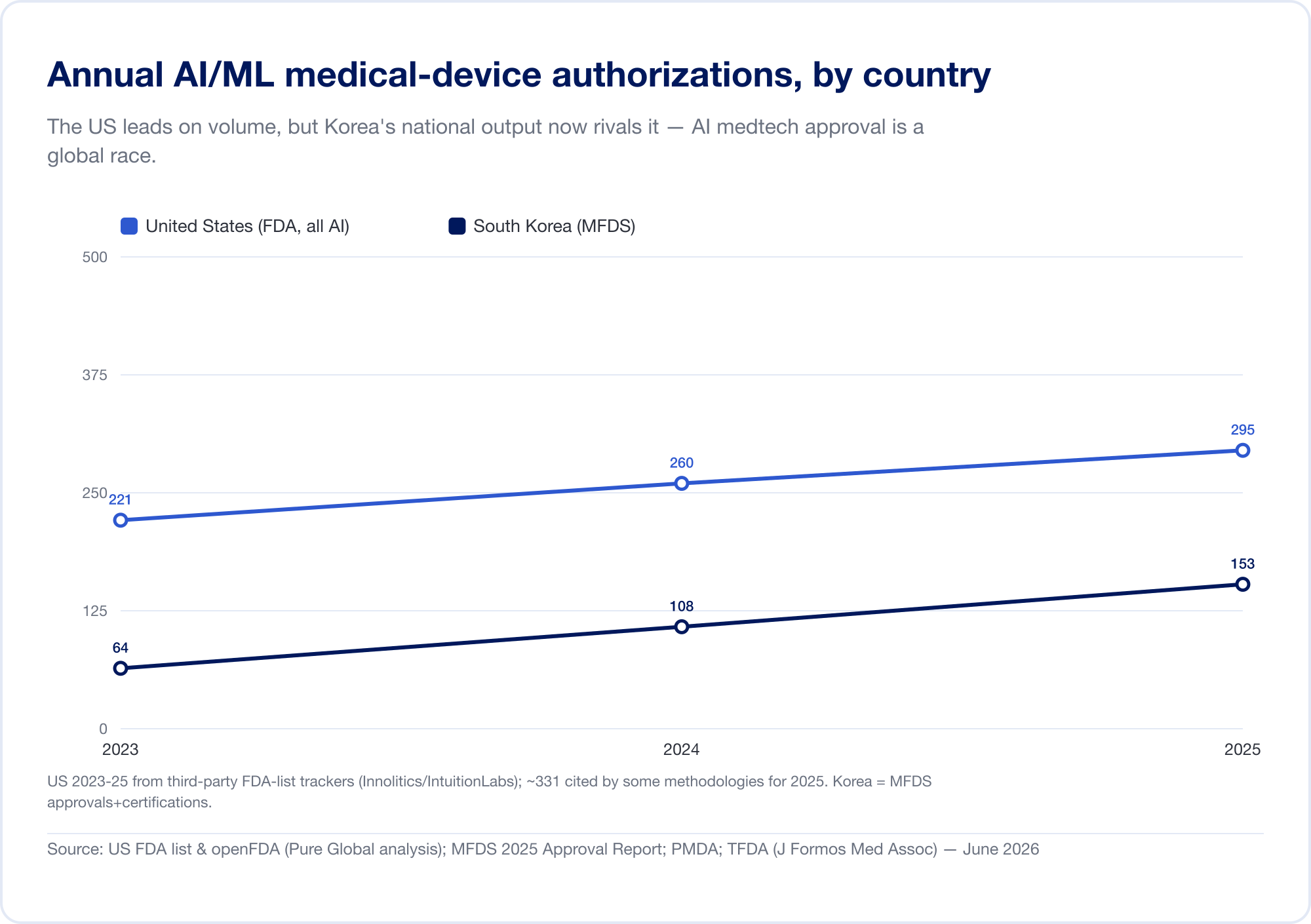
Die Implikation ist direkt: Ein brillanter Algorithmus, der in Tel Aviv, Seoul oder Shanghai entwickelt wurde, muss Patienten in Märkten erreichen, die jeweils eine andere regulatorische Sprache sprechen. Innovation ist global; Die Zustimmung ist hartnäckig lokal.
Wie groß ist der Markt? Es hängt ganz davon ab, was Sie zählen
Schätzungen zur Marktgröße von AI im Gesundheitswesen umfassen eine Größenordnung, da Analysten die Grenze an völlig unterschiedlichen Stellen ziehen. Die Breite „AI im Gesundheitswesen“ Die Kategorie „Medikamentenforschung“, „Verwaltungsautomatisierung“, „Ambient Scribes“ und mehr wird prognostiziert 187,7 Milliarden US-Dollar bis 2030 bei einer CAGR von 38,5 % (Grand View-Forschung), wobei das aggressivste Haus bis 2034 über 1 Billion US-Dollar prognostiziert (Fortune Business Insights). Die schmalere, regulierte Scheibe – AI in medizinischen Geräten – ist viel kleiner und glaubwürdiger: ungefähr 12,4 Milliarden US-Dollar im Jahr 2025 und 42,4 Milliarden US-Dollar im Jahr 2030 (Das Wirtschaftsforschungsunternehmen). Rein SaMD wird je nach Quelle auf etwa 5–25 Milliarden US-Dollar geschätzt (Mordor-Geheimdienst). Betrachten Sie jedes einzelne davon als einen Bereich, nicht als eine Tatsache.
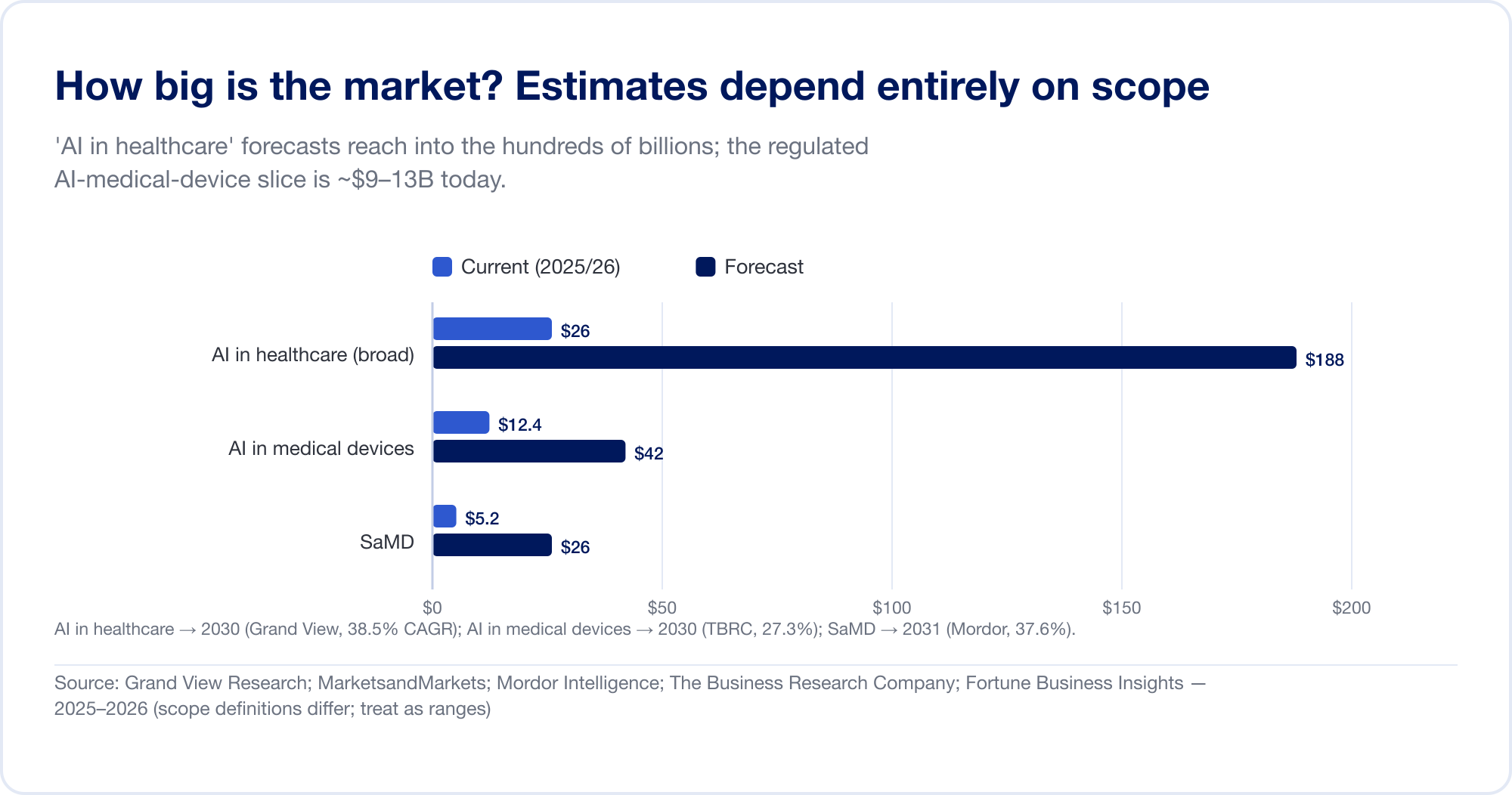
Die Hauptstadt erzählt eine klarere Geschichte. Die US-amerikanische Risikofinanzierung für digitale Gesundheit erholte sich 14,2 Milliarden US-Dollar im Jahr 2025 (+35 %), und zum ersten Mal eroberten AI-fähige Startups die Mehrheit – 54 % aller Dollars (Rock-Gesundheit). Öffentliche AI-SaMD-Namen verändern sich vom Wachstum zur Profitabilität: Tempus AI verzeichnete im Geschäftsjahr 2025 einen Umsatz von 1,3 Milliarden US-Dollar (+83 %) (Tempus); iRhythmus erreichte 747 Millionen US-Dollar und sein erstes GAAP-gewinnbringendes Quartal (iRhythmus); HeartFlow ging im August 2025 an die Börse und wuchs um 40 % auf 176 Mio. USD (HeartFlow); Koreas Lunit wuchs um 53 %, wobei 92 % des Umsatzes im Ausland erzielt wurden (Lunit). Unter Privatspielern Aidoc hat im April 2026 eine Serie-E-Finanzierung in Höhe von 150 Millionen US-Dollar eingesammelt, was einer Gesamtfinanzierung von über einer halben Milliarde US-Dollar entspricht (Aidoc).
Der Patentwettlauf hat einen anderen Anführer
Im Bereich des geistigen Eigentums ist der globale Wettbewerb am deutlichsten sichtbar. Die wegweisende Studie der WIPO zählte ungefähr 340.000 AI-bezogene Patentanmeldungen, wobei die Lebens- und Medizinwissenschaften das drittgrößte Anwendungsfeld darstellen (WIPO). Doch seitdem hat sich die Geographie gewandelt: Im Jahrzehnt bis 2023 China meldete 38.210 generative AI-Erfindungen an – mehr als der Rest der Welt zusammen – gegenüber 6.276 aus den Vereinigten Staaten (WIPO). Chinas Patentamt hat am letzten Tag des Jahres 2024 spezielle AI-Patentprüfungsrichtlinien herausgegeben (CNIPA). Die Pipeline an medizinischen AI wird überproportional aus Asien gefüllt – was eine Multi-Market-Access-Strategie zur Frage stellt wann, nicht wenn, für einen wachsenden Anteil der Entwickler weltweit.
Der blinde Fleck: Sie können SaMD nicht in den Handelsdaten sehen
Eine analytische Warnung, die die Denkweise über diesen Markt neu definiert. Traditionelle medizintechnische Marktinformationen stützen sich auf Import- und Zolldaten – aber reines SaMD ist für ihn unsichtbar. Software, die per Cloud-Download oder App-Store bereitgestellt wird, überschreitet keine physische Grenze, generiert keine HS-Code-Zollanmeldung und wird nicht in der Warenhandelsstatistik erfasst. Die internationalen Standardsetzer sagen es direkt: „Datenströme, die nicht direkt monetarisiert werden, werden in den aktuellen statistischen Standards im Allgemeinen nicht als Handelsströme betrachtet.“ (OECD-IWF-WTO), und das WTO-Moratorium für Zölle für elektronische Übermittlungen gilt seit 1998 (UNCTAD). AI-eingebettete Hardware – ein AI-fähiger CT-Scanner – bewegt sich als Ware und erscheint in den Daten; Bei einem 510(k)-zertifizierten Cloud-Algorithmus ist dies nicht der Fall. Das Ergebnis: Konventionelle Handelsanalyse mit System unterzählt AI-Software und übergewichtete Hardware. Der einzige verlässliche Fußabdruck der globalen Verbreitung eines SaMD ist sein Anmeldungen – Das ist genau die Linse, die dieser Bericht verwendet.
Die Sicherheitsabrechnung
Hinter jeder Verschärfung der Regeln steckt eine Fülle von Beweisen dafür, dass AI in der Medizin auf eine Weise versagen kann, die bei herkömmlichen Geräten nicht der Fall ist. Insbesondere drei Erkenntnisse haben die Denkweise der Regulierungsbehörden verändert.
Voreingenommenheit, die sich in einfachen Daten verbirgt. Ein Wahrzeichen New England Journal of Medicine Eine Studie ergab, dass Pulsoximeter – allgegenwärtig und zunehmend mit Algorithmen gekoppelt – gefährlich niedrige Blutsauerstoffwerte (okkulte Hypoxämie) übersehen 11,7 % der schwarzen Patienten gegenüber 3,6 % der weißen Patienten, eine ungefähr dreifache Disparität über etwa 50.000 gepaarte Messwerte (NEJM, 2020). Das FDA veröffentlichte im Februar 2021 eine Sicherheitsmitteilung und bis Januar 2025 einen Leitlinienentwurf, der eine vielfältigere Validierung über Hauttöne hinweg fordert (FDA). Die Lektion verallgemeinerte: Ein Modell, das an einer nicht repräsentativen Bevölkerung trainiert wird, kann diese Voreingenommenheit stillschweigend in jedes Krankenhaus übertragen, das sie einsetzt.
Bestätigung, die den Kontakt mit der Realität nicht übersteht. Das Epic-Sepsis-Modell – ein proprietäres Vorhersagetool, das in Hunderten US-Krankenhäusern eingesetzt wird – wurde an 38.455 Krankenhauseinweisungen extern validiert und erzielte eine Bewertung Fläche unter der Kurve von 0,63, weit unter den vom Verkäufer angegebenen 0,76–0,83. Es wurden 67 % der Sepsis-Fälle übersehen, während bei 18 % aller Patienten Alarme ausgelöst wurden (JAMA Innere Medizin, 2021). Ein Modell kann in großem Maßstab „in Produktion“ sein und dennoch nicht wie beworben funktionieren.
Erinnert sich an diesen Cluster direkt nach der Freigabe. Eine Johns Hopkins/Yale-Analyse von FDA-autorisierten AI-Geräten aus dem Jahr 2025 ergab dies 43,4 % der AI-Geräterückrufe erfolgten innerhalb der ersten 12 Monate nach der Freigabe – etwa doppelt so viel wie bei 510(k)-Geräten insgesamt (JAMA Gesundheitsforum, 2025). Eine parallele Studie ergab, dass Rückrufe sich auf Geräte konzentrierten, für die keine veröffentlichten klinischen Studien vorliegen (JAMA Network Open, 2025). Der Kontext ist wichtig: ungefähr 97 % der AI/ML-Geräte werden über den 510(k)-Weg gelöscht, das keine prospektiven Tests am Menschen erfordert – daher hängt ein großer Teil von der Wachsamkeit nach dem Inverkehrbringen ab.
Die Sicherheitsabwägung: Warum die Regulierungsbehörden AI verschärfen
Voreingenommenheit, Validierungslücken und frühe Rückrufe sind der Beweis für den Übergang zur Lebenszyklusüberwachung.
| Finden | Abbildung | Quelle |
|---|---|---|
| Okkulte Hypoxämie mit Pulsoximeter, schwarze vs. weiße Patienten | 11,7 % vs. 3,6 % (~3x) | NEJM, Dezember 2020 |
| Externe AUC des epischen Sepsis-Modells (vs. 0,76–0,83 behauptet) | 0,63; 67 % der Sepsisfälle wurden übersehen | JAMA-Praktikant. Med., Juni 2021 |
| AI-Geräterückrufe innerhalb von 12 Monaten nach Freigabe | 43,4 % (~2x alle 510(k)s) | JAMA Gesundheitsforum, 2025 |
| AI-Geräte zurückgerufen (903 untersucht) | 4,8 %, konzentriert auf diejenigen, für die keine klinischen Studien vorliegen | JAMA Network Open, 2025 |
| AI-Geräte über 510(k) freigegeben (keine prospektiven Tests erforderlich) | ~97% | Analysen der FDA-Liste, 2025 |
Hinzu kommt das Problem von Datensatzverschiebung – eingesetzte Modelle verschlechtern sich stillschweigend, wenn sich die Patientenpopulation, Scanner oder Codierungssysteme um sie herum ändern – und Sie haben die Begründung für den gesamten modernen Apparat: Gute maschinelle Lernpraxis, vorab festgelegte Änderungskontrollpläne und obligatorische Leistungsüberwachung in der Praxis. Die Regulierungsbehörden verschärfen nicht, weil AI nicht funktioniert. Sie verschärfen sich, weil es funktioniert bis es leiser wird.
Die globale Regulierungskarte
Dies ist der Referenzkern des Berichts: Wie die wichtigsten Gerichtsbarkeiten AI SaMD ab Mitte 2026 tatsächlich klassifizieren, überprüfen und überwachen. Die zu beobachtende Durchgangslinie ist Einstufung (in welche Risikoklasse die Software fällt) und Änderungskontrolle (Was passiert, wenn der Algorithmus aktualisiert wird). Eine konsolidierte Vergleichsmatrix folgt dem regionalen Detail.
Wie 15 Gerichtsbarkeiten AI als Medizinprodukt regulieren (2026)
Dieselbe Software, fünfzehn Antworten: Klassifizierung, AI-spezifische Anleitung und Änderungskontrollregeln unterscheiden sich von Markt zu Markt.
| Gerichtsstand | Wo die meisten AI SaMD landen | Spezielle AI/SaMD-Anleitung | Änderungskontroll-/adaptiver AI-Mechanismus | Vertrauen auf ausländische Zulassung |
|---|---|---|---|---|
| Vereinigte Staaten (FDA) | Klasse II (510(k)/De Novo) | Ja – PCCP-Finale 2024; Lebenszyklusentwurf 2025 | PCCP (Änderungen vorab autorisieren, keine neue Einreichung) | Nein (eigene Rezension; prädikatbasiert) |
| Europäische Union | Klasse IIa+ (MDR Regel 11) + AI Handeln mit hohem Risiko | MDR + AI Act + MDCG 2025-6 | Wesentliche Änderung → erneute Überprüfung der benannten Stelle + AI-Gesetz | Nein (CE-Konformitätsbewertung) |
| Vereinigtes Königreich (MHRA) | Klasse IIa+ (UK MDR 2002) | Software- und AI-Änderungsprogramm; AI Luftschleuse | PCCP geplant (gesetzliches Instrument) | CE wird in GB bis 2028/2030 akzeptiert |
| Kanada (Gesundheit Kanada) | Klasse II–IV | Ja – ML-Leitlinien endgültig, April 2026 | PCCP | MDSAP für QMS; keine vollständige Produktvertrauen |
| Australien (TGA) | Klasse IIa–III | AI-Überprüfung 2024 (14 Ergebnisse) | In Entwicklung | Vergleichbare-Übersee-Regulierungsroute |
| Japan (PMDA/MHLW) | Klasse II–III | DASH für SaMD; SaMD-Anleitung | IDATEN (PACMP) vorab vereinbarte Änderungen | Ausländische klinische Daten werden akzeptiert; MDSAP |
| Südkorea (MFDS) | Klasse 2–3 | Ja – inkl. Weltweit erste generative AI-Richtlinie | DMPA vorab genehmigte Änderungspläne | Begrenzt; eigene Rezension |
| Singapur (HSA) | Klasse B–D | Ja – GL-04-R4 (2025), AI-MD-Lebenszyklus | Änderungsbenachrichtigung | Ja – 5 Referenzagenturen (~98 % gekürzt) |
| China (NMPA) | Klasse III (Entscheidungssoftware) | Ja – CMDE AI Prinzipien + Klassifizierungskatalog | Ausgliederung nur, wenn der Kernalgorithmus unverändert bleibt | Nein (Vertreter im Land; Typprüfung) |
| Indien (CDSCO) | Klasse A–D | Nur Entwurf (Okt 2025) | Algorithmusänderungsprotokoll (vorgeschlagen) | Die Zulassung des Referenzlandes erleichtert die Klasse C/D |
| Taiwan (TFDA) | Klasse II–III | Ja – CADe/CADx + PCCP-Entwurfsanleitung | PCCP-Leitfaden (2024) | Lokale Leistungsbewertung wird hervorgehoben |
| Brasilien (ANVISA) | Klasse II–IV (Regel 11) | RDC 657 (SaMD); Keine dedizierte AI-Regel | Vollständige Änderungsregistrierung | Ja – IN 290/2024 (Klasse III/IV, 4 Agenturen) |
| Mexiko (COFEPRIS) | Klasse I–III | Allgemeine Geräteregeln | Neuanmeldung | Ja – abgekürzter Pfad (IMDRF + MDSAP) |
| Saudi-Arabien (SFDA) | Klasse A–D | Ja – MDS-G010 (früh; als erstes „durchsetzbar“ bezeichnet) | Änderungsmitteilung über GHAD | Nur unterstützend; lokale Validierung erforderlich |
| Vereinigte Arabische Emirate (EDE) | Klasse I–III | Allgemeine Geräteregeln | Neuanmeldung | Ja – erkennt CE/FDA |
Quelle: US FDA, EU MDR/AI Act, MHRA, Health Canada, TGA, PMDA/MHLW, MFDS, HSA, NMPA, CDSCO, TFDA, ANVISA, COFEPRIS, SFDA, EDE – Pure Global-Analyse, Juni 2026.
Die Vereinigten Staaten sind der Maßstab und am geschäftigsten
Die USA betreiben das weltweit aktivste AI-Geräteregime, das vom Center for Devices and Radiological Health des FDA und seinem Digital Health Center of Excellence verwaltet wird. Es gibt kein spezielles „AI-Statut“; AI-Funktionen, die der Gerätedefinition entsprechen, werden über drei Wege als SaMD reguliert: 510(k) Freigabe (Nachweis der „wesentlichen Gleichwertigkeit“ mit einem Prädikatgerät), De Novo Klassifizierung (für neuartige Geräte mit geringem bis mittlerem Risiko ohne Prädikat) und PMA (Zulassung vor dem Inverkehrbringen für die höchste Risikoklasse III). Die überwältigende Mehrheit der AI-Geräte – etwa 97% — Eingabe über 510(k); nur ein paar Dutzend haben De Novo verwendet und eine Handvoll PMA (Analyse der FDA-Liste). Das Wahrzeichen von De Novo war IDx-DR im Jahr 2018.
Die entscheidende jüngste Entwicklung ist die Vorab festgelegter Änderungskontrollplan (PCCP), fertiggestellt im Dezember 2024 (FDA). Ein PCCP ermöglicht es einem Hersteller, eine Reihe zukünftiger Modellmodifikationen vorab zu spezifizieren und zu genehmigen – und die eigenen Worte der Behörde spiegeln den Wert wider: Der FDA überprüft den PCCP „um die anhaltende Sicherheit und Wirksamkeit des Geräts zu gewährleisten, ohne dass für die Implementierung jeder Änderung zusätzliche Marketinganträge erforderlich sind.“ Im Januar 2025 ging FDA noch einen Schritt weiter und veröffentlichte einen umfassenden Leitlinienentwurf zum gesamten Produktlebenszyklus von AI-fähiger Gerätesoftware. Kurz gesagt, die Haltung der USA: schnell, prädikatgesteuert, bildlastig und jetzt auf die Überwachung des Lebenszyklus ausgerichtet.
Europäische Union – zwei Regime gestapelt auf einem Gerät
Der EU ist der schwierigste große Markt für AI und SaMD, da jetzt zwei Regulierungssysteme gleichzeitig gelten. Erstens, die Verordnung über Medizinprodukte (MDR). Seine Softwareklassifizierungsregel – Regel 11 – ist berüchtigt. Lesen Sie es direkt: „Software, die dazu bestimmt ist, Informationen bereitzustellen, die zur Entscheidungsfindung zu diagnostischen oder therapeutischen Zwecken verwendet werden, wird in die Klasse IIa eingestuft.“ eskaliert zu Klasse III wenn eine falsche Entscheidung zum Tod oder zu einer irreversiblen Verschlechterung führen könnte, oder Klasse IIb für eine schwerwiegende Verschlechterung (MDR, Anhang VIII, über EUR-Lex). Nach den alten Richtlinien wurde die meiste eigenständige Software selbst als Klasse I zertifiziert, ohne dass Dritte beteiligt waren. Regel 11 hat fast alle diagnostischen und therapeutischen SaMD in Klasse IIa oder höher gehoben, was eine Konformitätsbewertung durch eine Benannte Stelle, ein Qualitätsmanagement nach ISO 13485 und eine klinische Bewertung erzwingt. Unser eigener Scan der EUDAMED-Datenbank hat AI-Schlüsselwortgeräte gefunden, die sich auf Klasse IIa konzentrieren – genau den Fußabdruck von Regel 11.
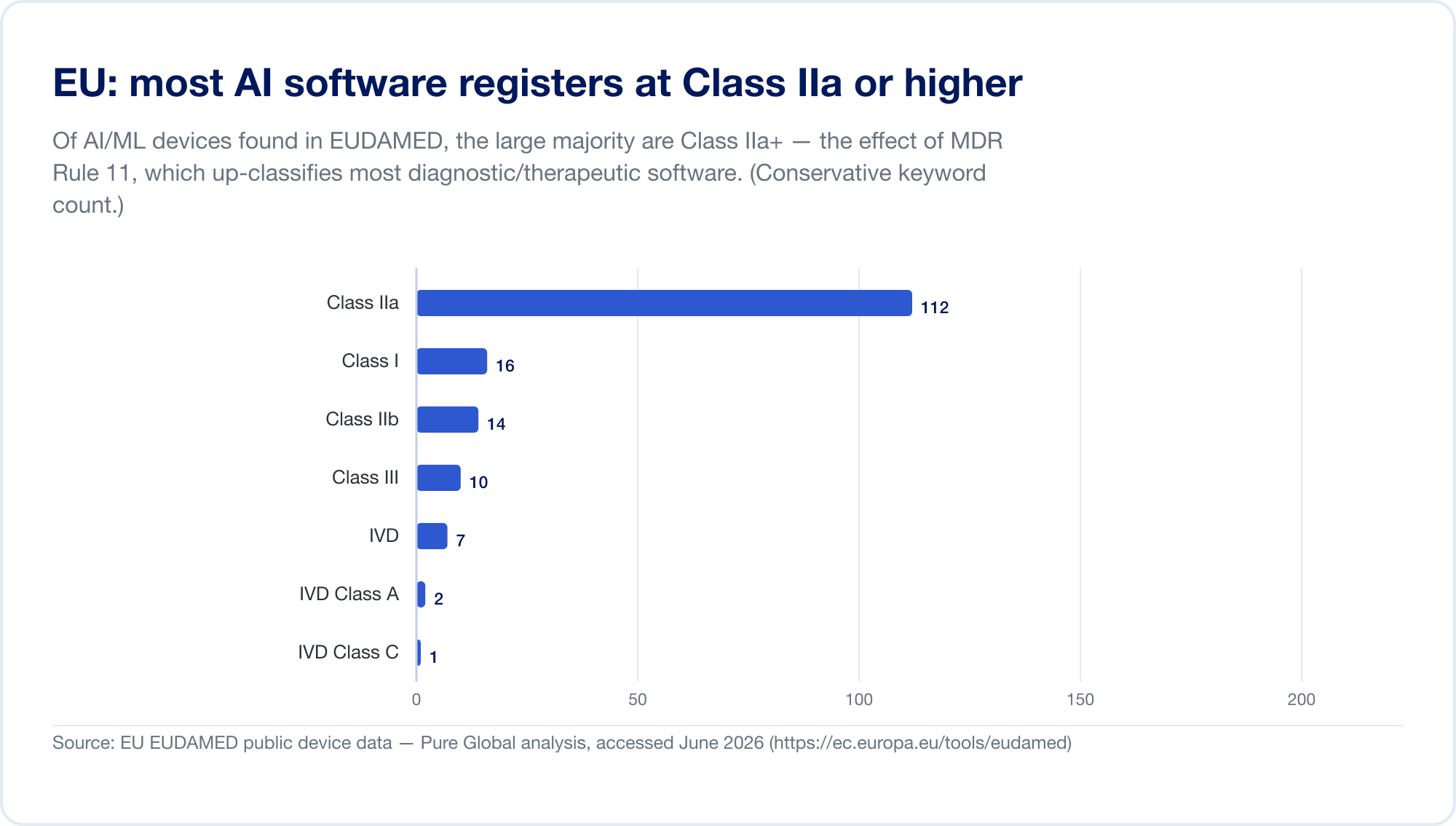
Dann, darüber geschichtet, das EU AI Act (Verordnung 2024/1689), in Kraft seit 1. August 2024. Gemäß Artikel 6 Absatz 1 gilt ein AI-System als „hochriskant“, wenn es sich um ein Produkt (oder eine Sicherheitskomponente davon) handelt, das bereits eine Konformitätsbewertung durch Dritte erfordert – was im Wesentlichen alle medizinischen Geräte der Klasse IIa+ AI erfasst. Verpflichtungen mit hohem Risiko treten schrittweise bis zum 2. August 2026 in Kraft, wobei das Datum für AI, das in MDR/IVDR-Geräten eingebettet ist, auf den 2. August 2027 festgelegt ist (Ein „Digital Omnibus“-Vorschlag vom November 2025 könnte dies auf 2028 verschieben – betrachten Sie die Daten als verschiebend) (künstlicheintelligenzact.eu). Die Strafen betragen bis zu 35 Millionen Euro oder 7 % des weltweiten Umsatzes (Artikel 99). Um die Überschneidung zu klären, haben das MDCG und das neue europäische AI-Board im Juni 2025 eine gemeinsame FAQ, MDCG 2025-6, herausgegeben (Europäische Kommission).
Die verbindliche Einschränkung ist die Kapazität. Die Zahl der MDR-Benannten Stellen sank von etwa 80–96 gemäß den Richtlinien auf etwa 50, wobei nur ~17–19 unter dem IVDR ausgewiesen sind; Die MDR-Zertifizierung dauert jetzt Durchschnittlich 13–18 Monate, ungefähr das Doppelte der Norm vor MDR (MedTech Europa). Für AI SaMD konkurriert jedes Gerät um knappe Plätze für benannte Stellen – und nach 2027/28 muss auch die AI Act-Konformitätsbewertung durchgeführt werden.
Vereinigtes Königreich – divergierend, pragmatisch
Nach dem Brexit hat der MHRA einen bewusst innovationsfreundlichen Kurs eingeschlagen. Es ist Software und AI als Programm zur Änderung medizinischer Geräte (Roadmap veröffentlicht im Oktober 2022) umfasst elf Arbeitspakete, und die Agentur hat sich verpflichtet, PCCPs in den kommenden Vormarktregeln zuzulassen (MHRA). Es ist AI Luftschleuse Die regulatorische Sandbox – die erste ihrer Art für AI-Medizingeräte – führte im Jahr 2024 ein Pilotprojekt mit vier Projekten und eine Phase 2 mit sieben Technologien bis 2026 durch, wobei nun eine mehrjährige Finanzierung zugesagt ist (MHRA). Praktisch immer noch tragen etwa 90 % der Geräte auf dem britischen Markt das CE-Zeichen, das Großbritannien bis dahin akzeptieren wird 2028–2030; die MHRA-Konsultation Anfang 2026 zur Erkennung von CE-Markierungen auf unbestimmte Zeit (MHRA).
Kanada – das erste Land, das spezielle ML-Regeln finalisiert
Health Canada gehörte zu den ersten Regulierungsbehörden abgeschlossen Spezielle Leitlinien vor der Markteinführung von medizinischen Geräten, die maschinelles Lernen ermöglichen – erstmals im Jahr 2025 fertiggestellt und in überarbeiteter endgültiger Form veröffentlicht April 2026 – Abdeckung der Klassen II–IV, Übernahme der IMDRF-Schlüsselbegriffe und formelle Einführung des PCCP, damit autorisierte Änderungen keine neue Lizenzänderung auslösen (Gesundheit Kanada). Kanada ist Mitautor der grundlegenden Dokumente der drei Regulierungsbehörden – GMLP (2021), PCCP-Grundsätze (2023) und Transparenzgrundsätze (2024) – und verlangt seit 2019 eine MDSAP-Zertifizierung.
Australien – frühzeitig reformiert, Neukalibrierung für AI
Australiens TGA hat seine Softwareregeln bereits im Februar 2021 reformiert und Wellness-Apps mit geringem Risiko eingeführt und gleichzeitig Diagnosesoftware hochklassifiziert (aktive Therapiegeräte mit Diagnosefunktion wurden in Klasse III verschoben). Seine Konsultation 2024, Klärung und Stärkung der Regulierung von AI, zog über 600 Interessenvertreter an und brachte 14 Schlüsselergebnisse hervor, die derzeit ausgearbeitet werden (TGA). Der Vertrauensweg des TGA – die Akzeptanz „vergleichbarer ausländischer Regulierungsbehörden“ – ist ein wichtiger Beschleuniger, der weiter unten erläutert wird.
Japan – für die Iteration gebaut
Japan reguliert SaMD als „programmierte medizinische Geräte“ und verfügt wohl über den AI-Update-freundlichsten Mechanismus aller großen Märkte: IDATEN, in Kraft seit September 2020, ist Japans Version eines Post-Approval-Change-Management-Protokolls, das es Herstellern ermöglicht, Änderungen an häufig aktualisierten AI vorab zu vereinbaren (PMDA). Kombiniert mit dem DASH für SaMD Mit der Initiative und dem vorrangigen SAKIGAKE-Pfad hat Japan eine Infrastruktur für sich weiterentwickelnde Software aufgebaut – allerdings ist die Akzeptanz bescheiden: Im September 2025 standen nur 51 AI-basierte SaMD auf der PMDA-Liste.
Südkorea – der schnellste Aufsteiger
Korea ist das Herausragende. Über seine 153 AI-Autorisierungen im Jahr 2025 hinaus hat es einen eigens dafür geschaffenen Rechtsrahmen geschaffen: den Gesetz über digitale Medizinprodukte (DMPA), gültig ab Januar 2025, führt einen Änderungsmechanismus im PCCP-Stil und ein digitales QMS ein, das auf IMDRF-Arbeitselemente ausgerichtet ist (Emergo). Korea hat auch das herausgegeben weltweit erste Richtlinie für generativ-AI-Medizinprodukte im Januar 2025 und genehmigte sein erstes Gerät dieser Art im April 2026 – und leitet die Arbeitsgruppe IMDRF AI/ML. Wenn Sie sehen möchten, wohin die globale AI-Regulierung führt, schauen Sie sich Seoul an.
Singapur – der Knotenpunkt des Vertrauens
Singapurs HSA ist seiner Größe weit überlegen, da es am effizientesten ist Vertrauen Regime in Asien. Seine Software-Anleitung, GL-04 (Revision 4, Dezember 2025)deckt explizit maschinell lernende Geräte im gesamten Lebenszyklus ab und erfordert eine Änderungsbenachrichtigung, wenn sich die Leistung, die Eingaben oder die menschliche Aufsichtsebene eines AI-Modells ändern (HSA). Entscheidend ist, dass HSA fünf Referenzagenturen anerkennt (US, FDA, EU Notified Bodies, Health Canada, TGA, Japan MHLW). Schätzungen zufolge können etwa 98 % der Anwendungen eine verkürzte Route verwenden; Geräte mit zwei vorherigen Genehmigungen können sich über einen „sofortigen“ Weg in nur einer Stunde registrieren (Singapur MOH).
China – groß, individuell und anspruchsvoll
Chinas NMPA nimmt die Entscheidungsunterstützungssoftware AI ernst: Die CMDE-Überprüfungsrichtlinien 2021 und der Klassifizierungskatalog 2021–2022 ordnen Software, die eine Diagnose liefert oder die Behandlung steuert, der Klasse III zu, der höchsten Risikostufe. China genehmigte im Jahr 2020 sein erstes AI-Gerät der Klasse III und hatte bis Mitte 2025 ungefähr 154 AI-Medizingeräte erreicht (JMIR Medizinische Informatik). In einem Reformpaket vom Oktober 2025 verpflichtete sich der NMPA dazu „Vereinfachen Sie die Änderungsregistrierungsanforderungen für AI-betriebene medizinische Geräte, bei denen der Kernalgorithmus unverändert bleibt, die Algorithmusleistung jedoch optimiert wird.“ – ein echtes, aber knappes Zugeständnis im Vergleich zum US-amerikanischen PCCP (NMPA). China erfordert einen Vertreter im Land, lokale Typprüfungen und – für viele Geräte der Klasse III – lokale klinische Daten, was es zu einem der anspruchsvollsten Märkte in diesem Bericht macht.
Indien, Taiwan und der Rest des asiatisch-pazifischen Raums
Indiens CDSCO hat im Oktober 2025 einen Entwurf einer Richtlinie für Medizingerätesoftware herausgegeben, in dem ein „Algorithm Change Protocol“ für AI-Updates eingeführt wird. Es handelt sich jedoch immer noch um einen Entwurf ohne endgültige AI-Regel (CDSCO). Taiwans Im Gegensatz dazu verfügt TFDA über eines der umfangreichsten AI-Anleitungspakete überhaupt – spezielle technische CADe/CADx-Richtlinien und PCCP-Entwurfsanleitungen – und hat von 2020 bis 2024 166 AI/ML-Geräte lizenziert. Bei ASEAN ist das Muster Vertrauen plus Lokalisierung: Malaysia, Thailand, Vietnam, die Philippinen und Indonesien Behandeln Sie SaMD meist als allgemeines Gerät und stützen Sie sich auf Genehmigungen des Referenzlandes, wobei Vietnams Fast-Track ungewöhnlich breit ist (es akzeptiert sogar NMPA- und MFDS-Genehmigungen).
Lateinamerika, der Nahe Osten und Afrika
Brasiliens ANVISA importierte die Logik der EU im Großen und Ganzen: Ihre Regel 11 unter RDC 751/2022 spiegelt die EU wider und ordnet Entscheidungsunterstützungssoftware der Klasse II–IV zu, und RDC 657/2022 war die erste SaMD-spezifische Auflösung der Region. Ein ausländischer Hersteller kann keine brasilianische Registrierung besitzen – eine lokale Brasilianischer Registrierungsinhaber ist obligatorisch und nicht verzichtbar (Artixio). Mexikos COFEPRIS hat sein Vertrauenssystem im Jahr 2025 in einen einzigen abgekürzten Pfad umgestaltet, der alle IMDRF- und MDSAP-Mitglieder berücksichtigt, mit einem Ziel von 30 Arbeitstagen. Saudi-Arabiens SFDA veröffentlichte MDS-G010 (November 2022) – einen der ersten dedizierten AI/ML-Leitfäden für medizinische Geräte überhaupt, von einigen Beobachtern als der erste durchsetzbare bezeichnet (andere stufen ihn als unverbindlich ein) –, der einzigartigerweise vorschreibt: „Der Hersteller sollte die AI/ML-basierten medizinischen Geräte, die in anderen Gerichtsbarkeiten entwickelt und zugelassen wurden, vor Ort validieren.“ (SFDA) – eine Erinnerung daran, dass eine „Genehmigung durch das Referenzland“ nicht immer ausreicht. Die Vereinigte Arabische Emirate Zentralisierte Gerätezulassung im Rahmen einer neuen Emirates Drug Establishment im Januar 2025. Südafrikas SAHPRA veröffentlichte seine erste AI-Mitteilung im September 2025, hat jedoch noch nicht mit der Registrierung von Geräten begonnen, und zwar auf dem Kontinent Afrikanische Arzneimittelbehörde – 31 von 55 Staaten ratifiziert – deckt Geräte oder AI noch nicht ab.
Das ist der Kern des Problems: fünfzehn Gerichtsbarkeiten, fünfzehn Antworten. Dieselbe Software ist Klasse II in den USA, Klasse IIa+ und „hohes Risiko“ im EU, Klasse III in China, Klasse 2–3 in Korea und Klasse II–IV in Brasilien – jeweils mit eigenen Nachweis-, Sprach-, lokalen Inhaber- und Änderungskontrollanforderungen.
Was es kostet und wie lange es dauert
Die oben genannten Klassifizierungsunterschiede lassen sich direkt in Geld und Monate umrechnen. Die Schlagzeilen unten sind staatliche Gebühren und realistische Zeitpläne für ein Gerät mit höherem Risiko; Sie schließen die erheblichen Kosten für Tests, klinische Beweise, Übersetzungen und die Vertretung im Land aus, die die offizielle Gebühr oft in den Schatten stellen.
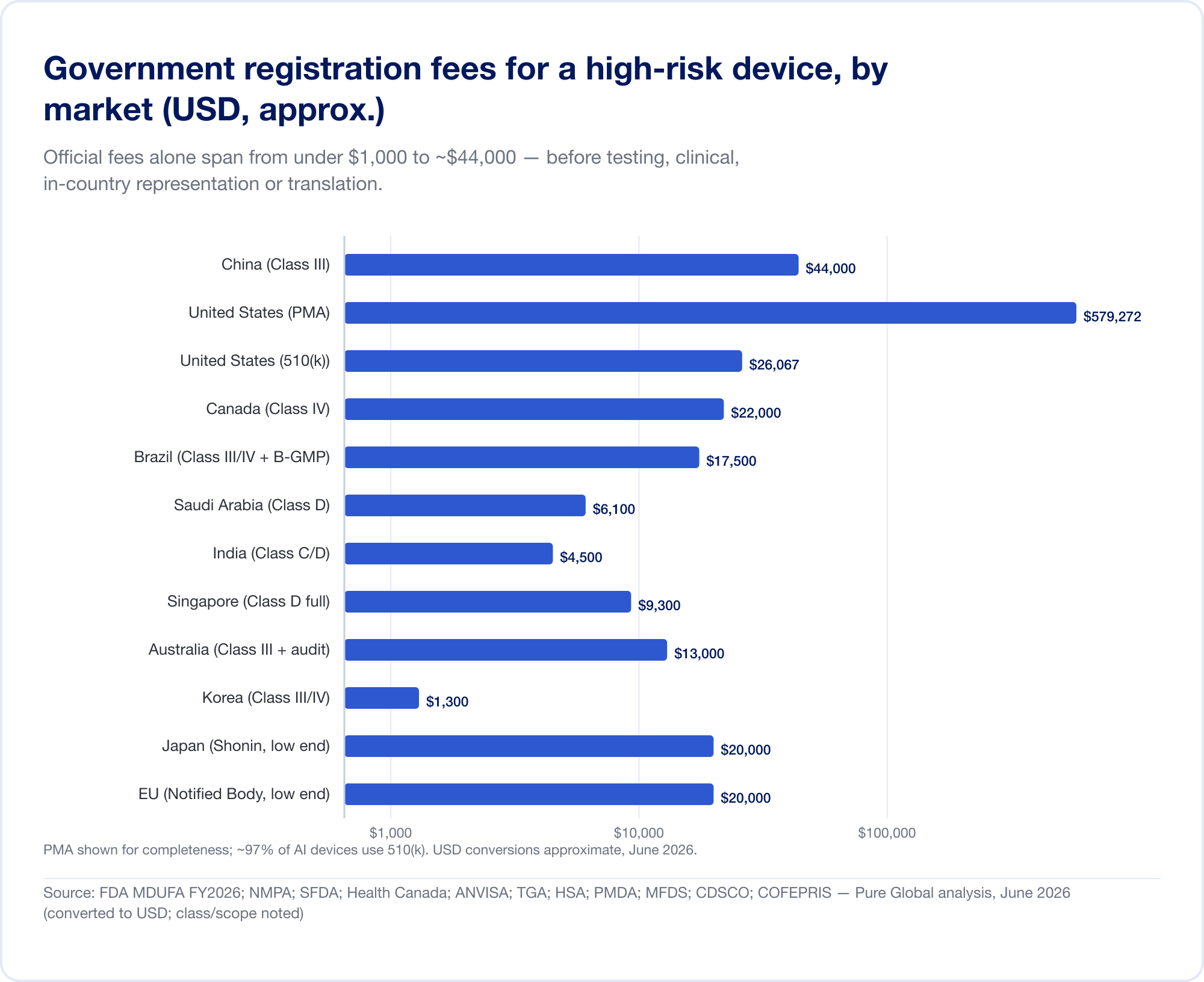
Die offizielle Gebühr ist die kleine Zahl
Allein die staatlichen Gebühren für eine Hochrisikoregistrierung liegen zwischen unter 1.000 USD und etwa 44.000 USD:
- Vereinigte Staaten – MDUFA-Gebühren für das Geschäftsjahr 2026 (überprüft auf FDA.gov): 510(k) $26,067 (Kleinunternehmen 6.517 $); De Novo $173,782; PMA $579,272; zuzüglich einer jährlichen Gründungsgebühr von $11,423 (FDA).
- China — NMPA-Registrierungsgebühren von ca 210.900 RMB (~30.000 $) für Klasse II und 308.800 RMB (~44.000 $) für Klasse III – die höchsten offiziellen Gebühren in diesem Bericht.
- Brasilien – ANVISA-Registrierung der Klasse III/IV von ~BRL 21.000 plus einer B-GMP-Zertifizierungsgebühr von BRL 72.804 für internationale Hersteller.
- Kanada — CAD der Klasse III $14,163, Klasse IV CAD $30,713 (April 2026).
- Saudi-Arabien — SFDA-Gebühren von 15.000–23.000 SAR nach Klasse.
- Indien — MD-15-Importlizenz von 3.000 $ pro Website + 1.500 $ pro Produkt für Klasse C/D.
- Singapur, Australien, Korea, Japan — Die offiziellen Gebühren sind vergleichsweise gering (oft unter 13.000 USD), aber der Beweis- und Überprüfungsaufwand variiert stark.
Zeit ist die teure Zahl
Der eigentliche Kostenfaktor ist der Kalender. Realistische Zeitpläne mit hohem Risiko reichen von etwa einem Monat (Kanada, Klasse III) bis 24 Monate im EU und vier bis fünf Jahre in China wenn eine lokale klinische Studie erforderlich ist:
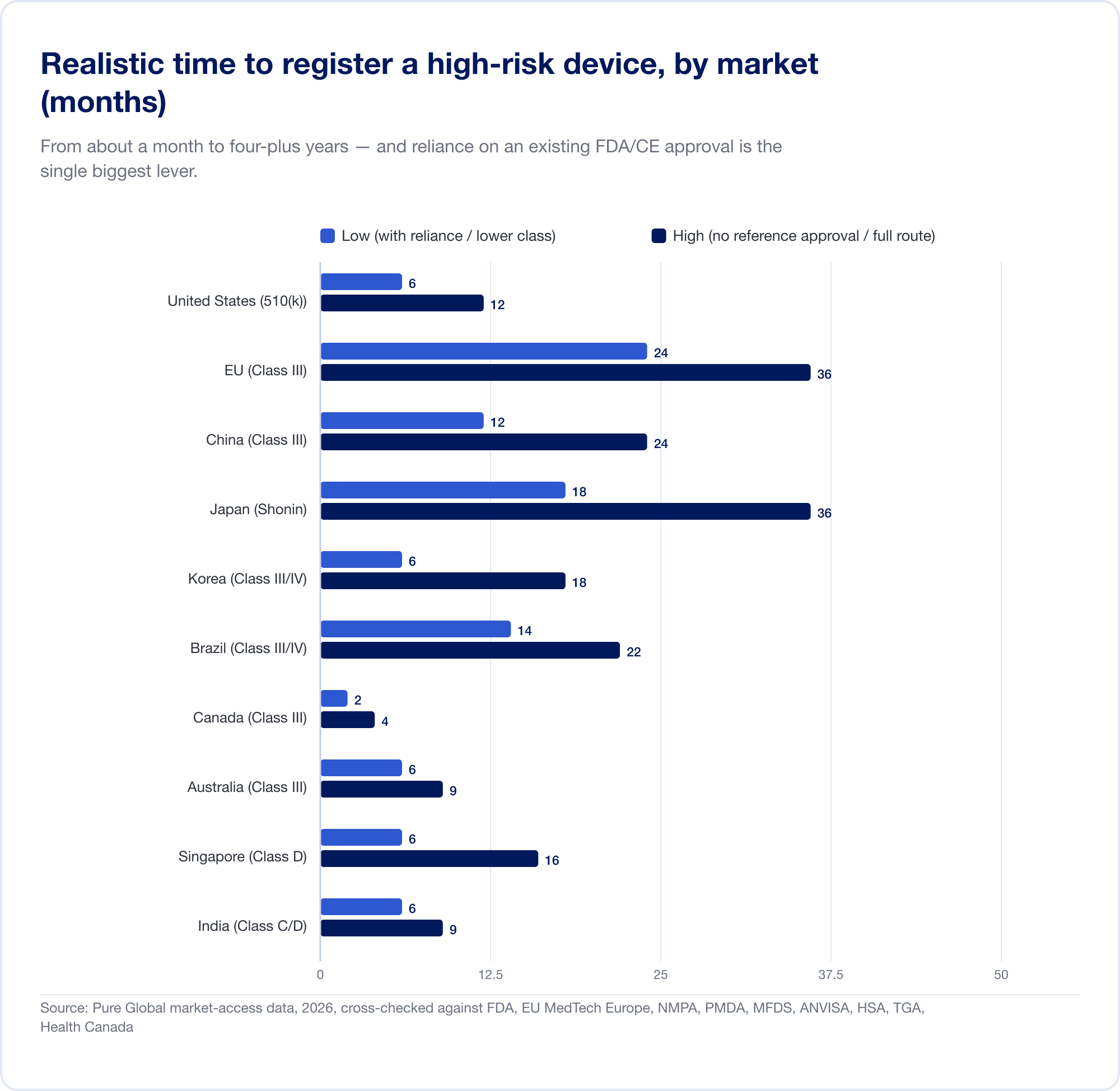
Das EU veranschaulicht, wie die Klassifizierung zu Kosten wird. Da Regel 11 eine ehemals nahezu kostenlose Selbsterklärung der Klasse I in eine Benannte Stelle der Klasse IIa+ umwandelt, beläuft sich das Gesamtprojekt CE für SaMD üblicherweise auf sechsstellige Beträge und 13–18 Monate bevor ein Zertifikat ausgestellt wird. Für ein Startup mit einer in Quartalen gemessenen Laufbahn ist das kein Einzelposten – es ist eine strategische Bedrohung.
Der Hebel: Vertrauen und die AI-spezifische Wendung
Diesen Zeitplänen steht das leistungsstärkste Instrument für den globalen Marktzugang gegenüber: Vertrauen. Die meisten Märkte außerhalb der USA, EU und China stützen sich auf eine Genehmigung, die Sie bereits besitzen. Unsere eigenen Marktdaten zeigen den Effekt deutlich: Ein risikoreiches SaMD, dessen Registrierung in Brasilien auf dem Standardweg etwa 8 Monate dauert, kann in Richtung fallen 6 Wochen wenn ein FDA oder eine andere Referenzgenehmigung über den optimierten Analysepfad von ANVISA genutzt wird.
Für AI SaMD gibt es eine bestimmte, wiederkehrende Wendung, die in den Kostentabellen nicht angezeigt wird. Wenn der Algorithmus aktualisiert wird – was AI ständig tut – stellt sich die Frage, ob diese Aktualisierung erforderlich ist neu Einreichung. Das US-amerikanische PCCP kann den Versand vorab festgelegter Updates ohne erneute Übermittlung zulassen und so die Kosten sparen 26.067 $ Gebühr plus 90–175-tägige Überprüfung für jedes vermiedene 510(k) und weitaus mehr für vermiedene PMA-Ergänzungsmittel. Über 10 % der AI-Geräte, die FDA im Jahr 2025 freigegeben hat, haben bereits ein PCCP eingebettet. Aber diese Ersparnisse gibt es nur in den USA – und das ist der Kern des gesamten Berichts.
Wie viel kostet die Registrierung von AI als Medizinprodukt in verschiedenen Ländern?
Die oben genannten staatlichen Gebühren betragen nur die Hälfte des Rechnungsbetrags. Die andere Hälfte ist die Facharbeit – das Erstellen des Dossiers, das Führen der lokalen Registrierung, das Beantworten aller Fragen der Behörde und das Einreichen jeder Verlängerung und Algorithmusänderung. Hier ist die Branche am undurchsichtigsten: Die meisten Regulierungsberatungsunternehmen rechnen stundenweise ab oder bieten für jede Einreichung ein separates Angebot an, sodass die tatsächlichen Multi-Markt-Kosten erst sichtbar werden, wenn die Änderungsaufträge eintreffen.
Pure Global ist das erste Marktzugangsunternehmen für medizinische Geräte, das eine einzige, pauschale Jahresgebühr pro Registrierung veröffentlicht. Von 2.000 USD pro Gerät, pro Markt und Jahr, dass eine Gebühr Dienstleistungen bündelt, die normalerweise stündlich abgerechnet werden – Vertretung im Land, Einreichung (auf Basis einer Referenzgenehmigung), Verlängerungen, Änderungen und die gesamte Korrespondenz mit der Gesundheitsbehörde. Keine Stundenzettel, keine Überraschungen per E-Mail.
Hier erfahren Sie genau, was es kostet, wenn Pure Global als Ihr lokaler Vertreter fungiert und eine AI-Registrierung für medizinische Geräte für jeden Markt vornimmt. (AI SaMD landet normalerweise in der höheren Risikoklasse, sodass in abgestuften Märkten der obere Wert gilt.)
Pure Global-Vertretung im Land – jährliche Pauschalgebühr pro AI-Gerät
Eine transparente Nummer pro Markt und Jahr – alles für die Registrierung inklusive.
| Markt | Lokale Rolle, die Pure Global bereitstellt | Pauschale Jahresgebühr (USD) |
|---|---|---|
| Vereinigte Staaten | FDA US Agent | $1,000 |
| Europäische Union | Autorisierter EU-Vertreter | $2,000 |
| Vereinigtes Königreich | UK Verantwortliche Person (UKRP) | $2,000 |
| Australien | TGA-Sponsor | $2,000 |
| Singapur | Registrant | 2.000 $ · 3.000 $ (Klasse C/D) |
| Malaysia | Autorisierter Vertreter | 2.000 $ · 3.000 $ (Klasse C/D) |
| Thailand | Autorisierter Vertreter | 2.000 $ · 3.000 $ (Klasse 3/4) |
| Indonesien | Autorisierter Vertreter | $2,000 |
| Vietnam | Inhaber der Marktzulassung | $2,000 |
| Hongkong | Lokale verantwortliche Person | 2.000 $ · 3.000 $ (Klasse III/IV) |
| Macau | Lizenzinhaber & Registrierung | 2.000 $ · 3.000 $ (Klasse III) |
| Brasilien | Brasilianischer Registrierungsinhaber (BRH) | 2.000 $ · 3.000 $ (Klasse III/IV) |
| Mexiko | Mexiko-Registrierungsinhaber | 2.000 $ · 3.000 $ (Klasse II/III) |
| Kolumbien | INVIMA-Vertreter | 2.000 $ · 3.000 $ (Klasse IIb/III) |
Pauschale Jahresgebühr für die Inlandsvertretung eines AI SaMD; Beinhaltet die Einreichung einer Referenzgenehmigung, Erneuerungen, Änderungen und behördliche Korrespondenz. Quelle: Pure Global Master-Preisliste, 2026 (pro Registrierung; es gelten Rabatte für Mehrfachregistrierungen und 3-Jahres-Verträge).
Einmalig Einreichung und Zusammenstellung Arbeiten – bei denen ein Markt ein vollständiges Dossier erstellen muss – werden ebenso transparent veröffentlicht: ein US-amerikanisches Die 510(k)-Zusammenstellung kostet zwischen 15.000 und 20.000 US-Dollar, wird eine technische Dokumentation oder ein CER-Projekt von EU aufgelistet nach Klasse (8.000–30.000 $), eine kanadische Registrierungszusammenstellung 3.000–25.000 US-Dollar pro Klasse, und eine Bestimmung des Regulierungspfads ist eine flache Sache $5,000. Jede Zahl wird im Voraus angegeben, niemals stundenweise.
Ein funktionierendes Beispiel – ein AI-Bildgebungsalgorithmus, vier Märkte. Nehmen Sie ein einzelnes AI-Radiologietool, das bereits mit dem FDA (Klasse II) und dem CE-Zeichen (Klasse IIb) freigegeben ist und das Sie ein Jahr lang in den Vereinigten Staaten, der Europäischen Union, Brasilien und Singapur am Leben halten möchten. Gesamtzahl der Inlandsvertretungen von Pure Global 1.000 $ (US) + 2.000 $ (EU) + 3.000 $ (Brasilien) + 3.000 $ (Singapur) = 9.000 $ für das Jahr – pauschal, einschließlich aller Erneuerungen, Änderungen und Autoritätsaustausche; Sie fügen nur dann einmalige Einreichungsarbeit hinzu, wenn ein Markt tatsächlich ein neues Dossier erfordert. Diese Vorhersehbarkeit ist der Punkt: Wenn die Regeln in jedem Markt unterschiedlich sind und sich Ihr Modell ständig ändert, ist das Letzte, was ein Hersteller braucht, ein Regulierungsgesetz, das dies auch tut.
Das Konvergenzparadoxon
Hier ist die scheinbar gute Nachricht. Unter dem Flickenteppich aus fünfzehn Gerichtsbarkeiten läuft eine mächtige Maschinerie von Harmonisierung. IMDRF bringt die Definitionen und die Grundsätze der Good Machine Learning Practice in Einklang. Die Einzelauditprogramm für Medizinprodukte (MDSAP) lässt ein einziges Qualitätssystem-Audit zufriedenstellen fünf Regulierungsbehörden gleichzeitig – die USA, Kanada, Brasilien, Japan und Australien (FDA). Und die Vertrauenswege verbreiten sich schnell: Singapur akzeptiert fünf Referenzagenturen und leitet etwa 98 % der Anträge über eine Kurzprüfung weiter; Brasilien, Mexiko, Australien, Malaysia, Vietnam und die Golfstaaten erkennen bis zu einem gewissen Grad ausländische Zulassungen an. Im Februar 2026 veröffentlichte IMDRF sogar eine globale Reliance Playbook die Praxis kodifizieren (IMDRF N89).
Welche Märkte akzeptieren welche ausländischen Zulassungen (Reliance Routes)
Eine FDA-Freigabe oder CE-Markierung ist ein Generalschlüssel für Dutzende von Märkten – jeder mit seinem eigenen Schloss.
| Markt | Anerkannte Referenzagenturen/Programme | Wirkung |
|---|---|---|
| Singapur (HSA) | US FDA, EU NB, Health Canada, TGA, Japan MHLW | Gekürzt/beschleunigt/sofort; ~98 % förderfähig |
| Brasilien (ANVISA) | TGA, Health Canada, US FDA, Japan MHLW (Klasse III/IV) | „Optimierte Analyse“ ~20–30 % schneller |
| Mexiko (COFEPRIS) | Alle IMDRF-Mitglieder + MDSAP-Teilnehmer | Verkürzter Weg, 30 Arbeitstage |
| Australien (TGA) | US FDA, Health Canada, MHLW/PMDA, EU NB, MDSAP | Verkürzte Konformitätsbewertung |
| Malaysia (MDA) | US FDA, Health Canada, TGA, EU CE, PMDA, HSA, Thai FDA | Verifizierungsroute (gekürzt) + MDSAP |
| Vietnam (MOH) | US FDA, EU, PMDA, TGA, Health Canada, MFDS, NMPA | Ungewöhnlich breiter SRA-Fast-Track |
| Saudi-Arabien (SFDA) | Nur unterstützend für FDA/CE | Eine vollständige Überprüfung der technischen Unterlagen ist noch erforderlich |
| Vereinigte Arabische Emirate (EDE) | CE, US FDA | Vertrauensbasierte Registrierung |
| MDSAP (ein Audit) | US, Kanada, Brasilien, Japan, Australien | Einzelnes QMS-Audit von allen fünf akzeptiert |
Quelle: HSA, ANVISA, COFEPRIS, TGA, MDA Malaysia, thailändisches FDA, Vietnam MOH, SFDA, EDE – Pure Global-Analyse, Juni 2026.
Für einen statisch Gerät ist dies transformativ: Eine starke Genehmigung – typischerweise FDA oder CE – wird zu einem Generalschlüssel, der Dutzende von Märkten mit verkürzter Geschwindigkeit und Kosten öffnet. Genau diesen Hebel soll ein gut geführtes Marktzugangsprogramm ausnutzen.
Und hier liegt das Paradoxon. Für adaptives AI, funktioniert der Hauptschlüssel genau an dem Punkt nicht mehr, der am wichtigsten ist – bei der Änderung der Kontrolle. Die Konvergenz liegt auf der Gerät; Die Divergenz liegt auf der AI. Stellen Sie sich dasselbe Modell für maschinelles Lernen vor, das seinen Algorithmus aktualisieren möchte:
- Im USA, ermöglicht ein vorab autorisiertes PCCP den Versand des Updates ohne erneute Übermittlung.
- Im EUEine „wesentliche“ Softwareänderung löst immer noch eine erneute Überprüfung durch die Benannte Stelle aus – und darüber hinaus eine separate AI Act-Konformitätsbewertung.
- In China, die Aktualisierung wird nur toleriert, wenn „der Kernalgorithmus unverändert bleibt“; Eine echte Umschulung bedeutet eine vollständige Änderungsregistrierung.
- In Korea, erlaubt das DMPA vorab genehmigte Änderungspläne – jedoch nur innerhalb vorab genehmigter Parameter.
Eine Analyse bringt es auf den Punkt: „Ein vom FDA autorisiertes PCCP erfüllt nicht die Verpflichtungen des EU AI Act und umgekehrt.“ (Berkley Lifesciences). Kein MDSAP-Audit und keine Vertrauensroute lösen dieses Problem. Ein AI-Entwickler, der in zehn Märkten Freigaben erhält, hat sich keinen Frieden erkauft; sie haben gekauft zehn verschiedene Change-Control-Verpflichtungen, jedes Mal ausgelöst, wenn sich das Modell verbessert. Für eine Technologie, deren gesamtes Wertversprechen darin besteht, dass sie immer besser wird, ist das eine strukturelle, sich verschärfende Belastung für den Erfolg – und sie trifft am stärksten die kleinen, schnell agierenden Teams, die die besten Modelle bauen.
Die nächste Grenze: generative AI- und Foundation-Modelle
Wenn das adaptive AI das System belastet, droht das generative AI es zu überfordern. Alles oben Genannte setzt ein Modell voraus, das für einen einzigen, genau definierten Verwendungszweck trainiert wurde – eine Blutung erkennen, eine Auswurffraktion messen, einen Knötchen markieren. Große Sprachmodelle und multimodale Basismodelle brechen diese Annahme auf drei Arten gleichzeitig: Sie sind es Allzweck (ein Modell, viele Einsatzmöglichkeiten), nicht deterministisch (die gleiche Eingabeaufforderung kann zu unterschiedlichen Antworten führen) und anfällig für Halluzination (sicher, fließend, falsch). Keine dieser Eigenschaften passt bequem in ein Framework, das auf einem festen Verwendungszweck und einer „gesperrten“ Referenzversion basiert.
Die Aufsichtsbehörden wissen es. Die WER gab im Januar 2024 den ersten globalen Leitfaden für große multimodale Modelle heraus und warnte insbesondere vor fabrizierten Ergebnissen, Automatisierungsverzerrungen und der Schwierigkeit, Systeme zu validieren, die auf Daten im Internetmaßstab trainiert wurden (WER). Der FDAs Der Digital Health Advisory Committee widmete seine Eröffnungssitzung im November 2024 den Herausforderungen des gesamten Produktlebenszyklus generativer AI-fähiger Geräte (FDA). Und Südkorea, bezeichnenderweise der erste, veröffentlichte im Januar 2025 die weltweit erste Richtlinie für generativ-AI-medizinische Geräte und genehmigte im April 2026 sein erstes derartiges Gerät.
Aber Beratung ist nicht dasselbe wie ein Weg zur Freigabe. Ab Mitte 2026 gehen die etablierten Routen – 510(k), De Novo, CE-Kennzeichnung – immer noch davon aus, dass es sich um ein Gerät handelt, das man festnageln, anhand eines festen Standards testen und einfrieren kann. Ein allgemeines klinisches LLM erfüllt keine dieser Voraussetzungen vollständig, weshalb die erste Welle von „generativen AI im Gesundheitswesen“ hauptsächlich als Verwaltungstools (Ambient Scribes, Dokumentationshilfen) auf den Markt gelangt ist, die die Gerätedefinition umgehen, und nicht als freigegebene Diagnosegeräte. Die regulatorischen Grenzen für autonome, generative klinische AI werden tatsächlich noch gezogen – und die Märkte, die sie zuerst ziehen (Korea heute; andere werden folgen), werden prägen, wie der Rest der Welt sie kopiert. Für Entwickler besteht die praktische Lektion darin, zu beobachten, wo die Grenzen gesetzt werden, und die Regulierungsstrategie, nicht nur das Produkt, für ein bewegliches Ziel zu entwerfen.
Das Playbook für den Marktzugang
Wenn das Problem Fragmentierung ist, ist die Antwort ein System. Über die oben genannten Muster hinweg entsteht ein wiederholbares Spielbuch, um ein AI-Medizingerät auf die Welt zu bringen – und dort zu behalten.
1. Klassifizieren Sie, bevor Sie Beweise erstellen. Dasselbe Produkt kann je nach Markt und Anspruch der Klasse II oder III angehören. Ordnen Sie die Klassifizierungsregeln der Zielmärkte zu zuerst, weil sie die klinischen und technischen Beweise vorschreiben, die Sie benötigen. Durch die Einordnung der Behauptung und der Beweise auf den strengsten Zielmarkt wird vermieden, dass das Dossier später erneut erstellt werden muss.
2. Gewinnen Sie eine starke Zustimmung des Ankers – und nutzen Sie dann das Vertrauen bewusst aus. Eine FDA-Freigabe oder CE-Marke ist weit mehr als einen Markt wert; Es ist der Ausweis, der verkürzte Routen in Singapur, Brasilien, Mexiko, Australien, am Golf und darüber hinaus freischaltet. Die Kunst ist zu wissen welche Anker, den jeder Zielmarkt erkennt, und Weiterleitung des Dossiers entsprechend. Saudi-Arabien, das selbst für im Ausland zugelassene AI eine lokale Validierung verlangt, ist eine Erinnerung daran, dass Vertrauen eine Karte und keine Decke ist.
3. Sorgen Sie für eine Vertretung im Land, wo es obligatorisch ist – und das ist an den meisten Orten der Fall. Ein ausländischer Hersteller kann keine eigene Registrierung in Brasilien (BRH), dem EU (autorisierter Vertreter), China (gesetzlicher Vertreter), Japan (MAH/DMAH), Saudi-Arabien, den Vereinigten Arabischen Emiraten, Indien und vielen anderen Ländern besitzen. In jedem Fall ist eine örtliche juristische Person erforderlich, die die Registrierung vornimmt und der Gesundheitsbehörde vorsteht. Es ist unpraktisch, dreißig solcher Einheiten aufzustellen; Durch die Auslagerung an einen einzigen Partner wird eine Skalierung möglich.
4. Behandeln Sie die Änderungskontrolle als einen erstklassigen, marktübergreifenden Arbeitsablauf. Dies ist die AI-spezifische Disziplin. Erstellen Sie ein US-PCCP, aber zeichnen Sie auch auf, wie jeder Markt damit umgeht gleich Aktualisieren Sie – und gestalten Sie den Veröffentlichungsrhythmus des Algorithmus entsprechend dem restriktivsten Regime, das für Ihr Unternehmen von Bedeutung ist. Der Lebenszyklusplan ist nun Teil des Marktzugangsplans.
5. Führen Sie es als einen zusammenhängenden Vorgang aus, nicht als dreißig getrennte Anmeldungen. Es fallen keine einmaligen Registrierungskosten an; Es ist die Koordination – Übersetzungen, lokale Inhaber, Erneuerungskalender, Änderungsbenachrichtigungen und Überwachung nach der Markteinführung über Dutzende von Systemen hinweg, die unabhängig voneinander agieren.
Ein typischer Ablauf in der Praxis. Für einen Imaging-AI-Entwickler läuft der Rollout oft so ab: Sichern Sie sich die FDA 510(k)-Freigabe oder die CE-Markierung als Anker; melden Sie gleichzeitig Ihren Heimatmarkt und einen wichtigen Markt (Singapur oder Australien) an, um frühzeitig Einnahmen zu erzielen. Verwenden Sie diese Genehmigungen, um die verkürzten Routen nach Brasilien, Mexiko und in die Golfstaaten zu öffnen; dann erobern Sie die aufwendigen, aber wertvollen Märkte – China und Japan — wo lokale Tests oder klinische Daten unvermeidbar sind und die Vorlaufzeiten am längsten sind. Durchgehend wird ein einziger Änderungskontrollplan zentral verwaltet und den Aktualisierungsregeln jedes Marktes zugeordnet, sodass eine Modellverbesserung überall dort abgelegt wird, wo sie sein muss, und nirgendwo, wo sie nicht sein muss. Die Reihenfolge ist nicht willkürlich; Es sequenziert den Cashflow, die Wiederverwendung von Beweismitteln und die Märkte für knappe Ressourcen bewusst.
Das ist die Arbeit Pure Global ist für Folgendes konzipiert: Vertretung im Land und AI-unterstützte regulatorische Umsetzung im gesamten Land Über 30 Märkte, bereitgestellt zu einer jährlichen Pauschalgebühr und nicht zum offenen Stundenmodell, das die Branche standardmäßig verwendet. Die Daten in diesem Bericht stammen aus FDA, EUDAMED, NMPA, PMDA, MFDS, ANVISA und Dutzenden nationaler Register sowie aus unserem eigenen Kosten- und Zeitplandatensatz für die einzelnen Märkte und sind dieselben Informationen, die wir verwenden, um die globale Einführung eines Kunden zu sequenzieren. Der Zweck einer so gründlichen Kartierung des Labyrinths besteht darin, einen Kunden schnell durch das Labyrinth führen zu können.
Fazit: Vier Dinge zum Mitnehmen
AI ist mittlerweile eine Mainstream-Gerätekategorie, und die Daten belegen dies. Von etwa 1 von 700 FDA-Freigaben im Jahr 2019 auf 1 von 28 im Jahr 2025, wobei allein in den USA mehr als 1.500 AI-Geräte zugelassen sind und nationale Programme in Korea, China, Taiwan und Japan schnell skalieren, hat sich AI SaMD von der Neuheit zur Norm entwickelt.
Jede Aufsichtsbehörde stuft es hoch, und die meisten stimmen bei der Überwachung des Lebenszyklus überein. Regel 11 im EU und Brasilien, Klasse III in China, hohes Risiko gemäß dem EU AI-Gesetz – diagnostisches und therapeutisches AI wird als schwerwiegend behandelt, und die gemeinsame Antwort ist die Kontrolle des gesamten Produktlebenszyklus, GMLP und vorgegebene Änderungspläne.
Reliance führt eine Genehmigungsreise durch – mit Ausnahme des AI-Teils. Die Harmonisierungsmechanismen (IMDRF, MDSAP, Vertrauensrouten) ermöglichen tatsächlich eine starke Ankergenehmigung, Dutzende von Märkten zu erschließen. Die Änderungskontrolle von adaptive-AI weicht jedoch stark voneinander ab, sodass eine einzelne Genehmigung nicht überall gültig bleibt, während sich das Modell weiterentwickelt. Diese Lücke ist die entscheidende operative Herausforderung in diesem Bereich.
Der Wettbewerbsvorteil liegt in der Registrierungsmaschine, nicht in der Registrierung. Wenn die Regeln in jedem Markt unterschiedlich sind und sich jedes Jahr ändern, liegt der dauerhafte Vorteil bei denen, die sich schnell und überall registrieren und jede Zulassung bei jeder Modellaktualisierung am Leben erhalten können – als ein vernetztes System.
Sprechen Sie mit uns
Wenn Sie ein AI-Medizingerät bauen oder skalieren und abwägen, welche Märkte in welcher Reihenfolge erschlossen werden sollen und wie jede Zulassung bei der Verbesserung Ihres Modells gültig bleiben soll, dann ist das genau das Problem, das wir lösen. Sprechen Sie mit Pure Global über einen Marktzugangsplan, der auf den oben genannten Daten basiert – oder erkunden Sie unsere marktspezifischen Registrierungsleitfäden, um tiefer auf ein einzelnes Land einzugehen.
Quellen
Oben im Text zitierte Regierungs- und Harmonisierungsstellen; Die wichtigsten Referenzen sind unten gruppiert. Alle Abbildungen sind datiert; Marktgröße und Prognosezahlen sind Schätzungen Dritter, deren Umfang variiert und als Spannen zu verstehen ist.
Definitionen, Rahmenbedingungen und Leitprinzipien
- IMDRF — SaMD: Schlüsseldefinitionen (N10, 2013); Risikokategorisierung (N12, 2014); ML-fähige Medizingeräte: Schlüsselbegriffe (N67, 2022); GMLP-Leitprinzipien (N88, 2025); Reliance Playbook (N89, 2026). imdrf.org
- US FDA — Software als Medizinprodukt (SaMD); Vorgeschlagener Regulierungsrahmen für Änderungen am AI/ML-basierten SaMD (2019); AI/ML-Aktionsplan (2021). fda.gov
- WER – Ethik und Governance von AI für die Gesundheit (2021); Regulatorische Überlegungen zu AI für die Gesundheit (2023); LMM-Anleitung (2024); Gute Vertrauenspraktiken, TRS 1033 Anhang 10 (2021). who.int
Vereinigte Staaten
- FDA — Künstliche Intelligenz-fähige medizinische Geräte Liste (Update Q1-2026, 1.524 Geräte; ~76 % Radiologie); Endgültige Leitlinien des PCCP (Dezember 2024); Beratender Ausschuss für digitale Gesundheit auf generativem AI (November 2024); MDUFA-Gebühren für das Geschäftsjahr 2026. fda.gov · The Imaging Wire (Radiologie-Aktienanalyse, 2026); Innolitics & IntuitionLabs (Clearance-Tracker, 2025–26).
Europäische Union & UK
- EUR-Lex – Verordnung (EU) 2017/745 (MDR), Anhang VIII Regel 11; Verordnung (EU) 2024/1689 (AI-Gesetz), Artikel 6, 99, 113. Europäische Kommission – MDCG 2019-11 Rev.1 & MDCG 2025-6. MedTech Europe & Team-NB (Kapazität der benannten Stelle). MHRA – Software- und AI-Änderungsprogramm; AI Luftschleuse; CE-Erkennungsanleitung. gov.uk
Kanada, Australien, Japan, Korea
- Gesundheit Kanada – Leitlinien vor der Markteinführung von ML-fähigen Medizinprodukten (April 2026). kanada.ca
- TGA – Softwarereformen (2021); Ergebnisse der AI-Konsultation (2024–26). tga.gov.au
- PMDA/MHLW – SaMD-Anleitung; DASH für SaMD; IDATEN. pmda.go.jp
- MFDS – Genehmigungsbericht 2025 (153 AI-Geräte); DMPA; generative-AI-Richtlinie. mfds.go.kr; bioin.or.kr
Asien-Pazifik, Lateinamerika, MEA
- HSA Singapur – GL-04-R4; Vertrauens-/Referenzagenturen. hsa.gov.sg; moh.gov.sg
- NMPA China – AI-Klassifizierung und Reform im Oktober 2025. nmpa.gov.cn · JMIR Medical Informatics (154 AIMDs, 2026).
- CDSCO India – Entwurf eines MD-Software-Leitfadens (Okt. 2025). TFDA Taiwan – CADe/CADx-Anleitung; J. Formos. Med. Assoc. (166 Lizenzen).
- ANVISA Brasilien – RDC 751/2022 (Regel 11), RDC 657/2022, IN 290/2024. gov.br/anvisa
- COFEPRIS Mexiko – Abbreviated Pathway (2025). SFDA Saudi-Arabien – MDS-G010 (2022). VAE EDE – Bundesgesetzesdekret 38/2024. SAHPRA Südafrika – AI-Kommunikation (2025).
Markt, Unternehmen, Patente, Sicherheit
- Marktgröße: Grand View Research; Märkte und Märkte; Das Wirtschaftsforschungsunternehmen; Mordor-Geheimdienst; Fortune Business Insights (2025–26).
- Finanzierung & Unternehmen: Rock Health (Digital-Health-Finanzierung 2025); CB-Einblicke; Einreichungen des Unternehmens für das Geschäftsjahr 2025 (Tempus AI, iRhythm, HeartFlow, Butterfly Network, Lunit, VUNO); Aidoc (Serie E).
- Patente: WIPO-Technologietrends (2019) und Generative AI-Patentlandschaft (2024); CNIPA.
- Sicherheit: NEJM (Pulsoximeter-Bias, 2020); JAMA Innere Medizin (Epic Sepsis Model, 2021); JAMA Health Forum & JAMA Network Open (AI erinnert sich, 2025); FDA Pulsoximeter-Anleitung.
- Handel: OECD-IWF-WTO Messung des digitalen Handels (2021); WTO; UNCTAD.
- Pure Global proprietärer Marktzugangskosten- und Zeitplandatensatz (2026); openFDA-, EUDAMED- und MFDS-Datenbankanalyse – Pure Global, Juni 2026.



Sprechen wir,
wo immer Sie sind.
Ob Sie weitere Informationen suchen oder bereit zur Zusammenarbeit sind: Wir begleiten Sie durch jeden Schritt des regulatorischen Prozesses.
Kontakt