
AI como dispositivo médico: el mapa global de regulación, registro y acceso al mercado
La inteligencia artificial es la clase de dispositivo médico de más rápido crecimiento en la historia y la más divergente en cuanto a regulaciones. El FDA de EE. UU. ahora enumera 1,524 dispositivos habilitados para AI; Corea del Sur autorizó 153 en un solo año. Sin embargo, el mismo software es Clase II en EE. UU., Clase IIa+ y de "alto riesgo" en EU, y Clase III en China, cada uno con sus propias reglas de evidencia, titular local y control de cambios. Este informe describe cómo cada regulador importante clasifica, aprueba y controla AI como dispositivo médico, con costos de registro, cronogramas y rutas de dependencia en más de 30 mercados, y el manual para llegar a ellos sin reconstruir el expediente cada vez.
Una guía de campo basada en evidencia sobre cómo los reguladores del mundo clasifican, aprueban y controlan la inteligencia artificial en la atención médica y cómo llegar a más de 30 mercados sin reconstruir el expediente cada vez.
TL;DR
La inteligencia artificial se ha convertido en la categoría de dispositivo médico de más rápido crecimiento en la historia. La lista pública de dispositivos habilitados para AI de la Administración de Alimentos y Medicamentos de EE. UU. alcanzó 1.524 autorizaciones para mediados de 2026, con aproximadamente tres cuartas partes en radiología (FDA). En nuestro propio análisis de la base de datos FDA 510(k), la participación de AI en todas las autorizaciones aumentó de aproximadamente 1 de cada 700 en 2019 a aproximadamente 1 de cada 28 en 2025. Sólo Corea del Sur autorizó 153 dispositivos AI en 2025 (MFDS).
Pero el mismo software que pasa por una agencia puede estancarse en la siguiente. He aquí el argumento de este informe, en cinco puntos:
- AI es un dispositivo y la mayor parte es "software como dispositivo médico" (SaMD). Cuando el software diagnostica, clasifica o recomienda un tratamiento, está regulado como un bisturí o un escáner: en docenas de jurisdicciones, cada una con sus propias reglas.
- El mundo entero lo clasifica. La Regla 11 del EU, la Regla 11 de Brasil, la Clase III predeterminada de China para el diagnóstico AI: el software de diagnóstico y terapéutico casi en todas partes cae en una clase de mayor riesgo que exige revisión de terceros.
- El AI adaptativo rompió el modelo de aprobación única. Un modelo que sigue aprendiendo cambios después de su lanzamiento, por lo que los reguladores inventaron nuevos mecanismos (el Plan de Control de Cambios Predeterminado de EE. UU., el IDATEN de Japón, el DMPA de Corea) que no coinciden entre sí.
- La confianza converge, AI diverge. Se supone que una red de vías de confianza y reconocimiento permitirá que una aprobación abra muchos mercados. Funciona, para dispositivos estáticos. Para adaptativo AI, "un PCCP autorizado por FDA no cumple con las obligaciones de la Ley EU AI, y viceversa" (Ciencias de la vida de Berkley).
- Así, los ganadores industrializan el registro en múltiples mercados. La ventaja competitiva ya no es una autorización única; es la máquina operativa que convierte una aprobación en treinta y mantiene cada una válida a medida que evoluciona el algoritmo.
Esta es la brecha que cierra Pure Global: representación en el país y ejecución regulatoria asistida por AI en más de 30 mercados, con una tarifa anual fija. El resto de este informe es el mapa.
Qué es realmente "AI como dispositivo médico"
Comience con la palabra que hace el trabajo pesado: dispositivo. Si un software está "destinado a ser utilizado en el diagnóstico, tratamiento o prevención de enfermedades", es un dispositivo médico en casi todos los sistemas legales del mundo, ya sea que se ejecute en un chip dentro de un escáner de resonancia magnética o como una aplicación en el navegador de un radiólogo. AI no tiene su propio estatuto en la mayoría de los países; hereda todo el aparato de la ley sobre dispositivos médicos.
La definición de ancla proviene del Foro Internacional de Reguladores de Dispositivos Médicos (IMDRF), el organismo donde las principales agencias armonizan su vocabulario. En su documento fundacional de 2013, IMDRF definió Software como dispositivo médico (SaMD) como "software destinado a ser utilizado para uno o más fines médicos que realiza estos fines sin ser parte de un dispositivo médico de hardware" (IMDRF N10). El FDA adoptó ese lenguaje palabra por palabra y establece una distinción de tres vías que es importante para todo lo posterior (FDA):
- SaMD - software que es el dispositivo médico (un algoritmo de clasificación de rayos X de tórax, un detector de retinopatía diabética). Aquí es donde vive la mayoría de los AI clínicos.
- SiMD — software en un dispositivo médico, integral al hardware (el firmware que ejecuta una bomba de infusión).
- Software utilizado para fabricar o mantener un dispositivo que vuelve a estar regulado de forma diferente.
Para AI específicamente, el documento de términos clave de 2022 de IMDRF define un Dispositivo médico habilitado para aprendizaje automático como "un dispositivo médico que utiliza el aprendizaje automático, en parte o en su totalidad, para lograr el propósito médico previsto" (IMDRF N67).
La distinción que lo rompe todo: bloqueado versus adaptativo
La regulación tradicional de dispositivos se basa en un simple trato: usted demuestra que un dispositivo es seguro y eficaz una vez, en un diseño fijo, y ese diseño luego permanece igual. AI rompe esa suposición. El fundamental documento de debate de 2019 del FDA trazó la línea con precisión. Un algoritmo "bloqueado" es aquel que "proporciona el mismo resultado cada vez que se le aplica la misma entrada y no cambia con el uso" — una tabla de consulta, un árbol de decisión, un clasificador congelado (FDA, 2019). Un algoritmo adaptativo o de aprendizaje continuo cambia después de su implementación.
Esa única propiedad (software que se mejora a sí mismo en el campo) es la razón por la que AI necesitaba una década de nueva regulación. Si el producto que aprobó en enero no es el producto que se ejecuta en un hospital en junio, ¿qué aprobó exactamente? Cada marco de este informe es, en el fondo, un intento de responder a esa pregunta, y la respuesta compartida es el ciclo de vida total del producto (TPLC): la supervisión de toda la vida útil del dispositivo, no sólo del momento de su autorización.
Cómo deciden los reguladores qué tan difícil es mirar
El marco de categorización de riesgos de IMDRF de 2014 estableció la lógica que el mundo sigue ahora: el escrutinio que un SaMD merece se escala con dos factores: la importancia de la información que proporciona (lo hace informar, conducir, o diagnosticar/tratar?) y la gravedad de la situación sanitaria (no grave, grave o crítico) (IMDRF N12). Una aplicación que sugiere estiramientos para el dolor de espalda y un algoritmo que señala una hemorragia cerebral no son el mismo animal regulador, y esta cuadrícula de dos ejes explica la razón.
el Organización Mundial de la Salud añadió el andamiaje ético. Su informe de 2021 Ética y gobernanza de la inteligencia artificial para la salud establecer seis principios: proteger la autonomía; promover el bienestar y la seguridad; garantizar la transparencia y la explicabilidad; fomentar la responsabilidad y la rendición de cuentas; garantizar la inclusión y la equidad; y promover un AI receptivo y sostenible (OMS). La OMS siguió con consideraciones regulatorias en 2023 y, en enero de 2024, la primera guía global dirigida directamente a AI generativo y grandes modelos multimodales (QUIÉN).
¿Cómo llegamos aquí?
Los dispositivos médicos AI no llegaron con un solo avance; se fueron acumulando, país tras país, a lo largo de aproximadamente una década. Los hitos a continuación muestran cómo una definición de 2013 se convirtió en reglas específicas del ciclo de vida en los cinco continentes.
Cronología global de la regulación de dispositivos médicos AI (2013-2026)
En apenas una década, AI SaMD pasó de una definición de IMDRF a reglas de ciclo de vida dedicadas en cinco continentes.
| Fecha | Hito |
|---|---|
| diciembre de 2013 | IMDRF N10 define "Software como dispositivo médico" (SaMD) |
| enero de 2017 | FDA aprueba Arterys: primera nube + herramienta clínica de aprendizaje profundo (510(k)) |
| abril de 2018 | FDA autoriza IDx-DR: primer diagnóstico AI autónomo (De Novo) |
| mayo 2018 | El MFDS de Corea aprueba VUNO Med-BoneAge, el primer dispositivo AI de Corea |
| diciembre de 2018 | El PMDA de Japón aprueba EndoBRAIN (Clase III): el primer AI SaMD de Japón |
| abril de 2019 | Documento de debate de FDA sobre modificaciones a AI/ML SaMD (bloqueado frente a adaptable) |
| 2020 | China NMPA aprueba DeepVessel FFR: primer dispositivo AI de Clase III; Japón lanza IDATEN + DASH para SaMD |
| enero de 2021 | FDA AI/Plan de Acción ML; Jun 2021 Seis principios éticos de la OMS |
| octubre de 2021 | GMLP: 10 principios rectores (FDA + Health Canada + UK MHRA) |
| 2022 | Saudi SFDA MDS-G010: primera guía dedicada para dispositivos AI (citada por algunos como la primera "aplicable"); Términos de IMDRF N67 ML; Brasil RDC 751/657 |
| agosto de 2024 | EU La Ley AI entra en vigor; Guía LMM (generative-AI) de la OMS (enero de 2024) |
| diciembre de 2024 | FDA finaliza la guía del Plan de control de cambios predeterminado (PCCP) |
| enero de 2025 | IMDRF N88 GMLP final; Corea publica la primera directriz sobre dispositivos generativos AI del mundo; Borrador de guía del ciclo de vida de FDA AI |
| febrero de 2026 | IMDRF N89 Manual de estrategias de confianza |
| abril de 2026 | Health Canada finaliza la guía sobre dispositivos de aprendizaje automático; Corea aprueba el primer dispositivo generativo AI |
Fuente: Compilado a partir de IMDRF, US FDA, EU, NMPA, PMDA, MFDS, SFDA, ANVISA, Health Canada y documentos primarios de la OMS: Pure Global, junio de 2026.
Algunos momentos merecen énfasis. en enero 2017, el FDA aprobó Arterys, la primera herramienta clínica que combina la computación en la nube y el aprendizaje profundo (Cable de noticias de relaciones públicas). Entonces, en abril 2018, llegó el punto de inflexión: el FDA autorizó IDx-DR, el primer AI autorizado a realizar un diagnóstico autónomamente, sin que ningún médico interprete el resultado: una prueba de detección de retinopatía diabética para atención primaria que alcanzó una sensibilidad del 87,2 % y una especificidad del 90,7 % en su ensayo fundamental (npj Medicina Digital). En cuestión de meses, Corea (VUNO Med-BoneAge, mayo de 2018) y Japón (EndoBRAIN, diciembre de 2018) aprobaron sus propias primicias; China siguió en 2020 con DeepVessel FFR, su primer dispositivo AI de Clase III (npj Medicina Digital).
El andamiaje regulatorio se puso al día entre 2021 y 2025: la trilateral Buenas prácticas de aprendizaje automático principios en octubre de 2021, el EU Ley AI entrando en vigor en agosto de 2024, la guía final del FDA sobre el Plan de control de cambios predeterminado en diciembre de 2024 y —como muestra de qué tan rápido se mueve la frontera— la primera directriz del mundo para dispositivos generativos AI, publicada por Corea en enero de 2025 (Biomundo). En febrero de 2026, IMDRF había publicado un Manual de estrategias de confianza global para ayudar a los reguladores a apoyarse en el trabajo de los demás (IMDRF N89).
Los datos: qué tan grandes, qué tan rápidos, dónde
La curva de holgura se está curvando bruscamente hacia arriba.
El mejor indicador de la penetración de AI en la medicina es la lista pública de FDA de dispositivos médicos habilitados por inteligencia artificial, que alcanzó 1.524 autorizaciones en su actualización más reciente (que refleja datos hasta el primer trimestre de 2026) (FDA). Para ver la velocidad, analizamos la base de datos subyacente FDA 510(k) directamente. Las autorizaciones de AI/ML identificadas aumentaron de un puñado a finales de la década de 2010 a 115 en 2025 - y, lo que es más revelador, AI surgió de 0,14% de todas las autorizaciones 510(k) en 2019 a 3,57% en 2025, un aumento de aproximadamente 25 veces en seis años.
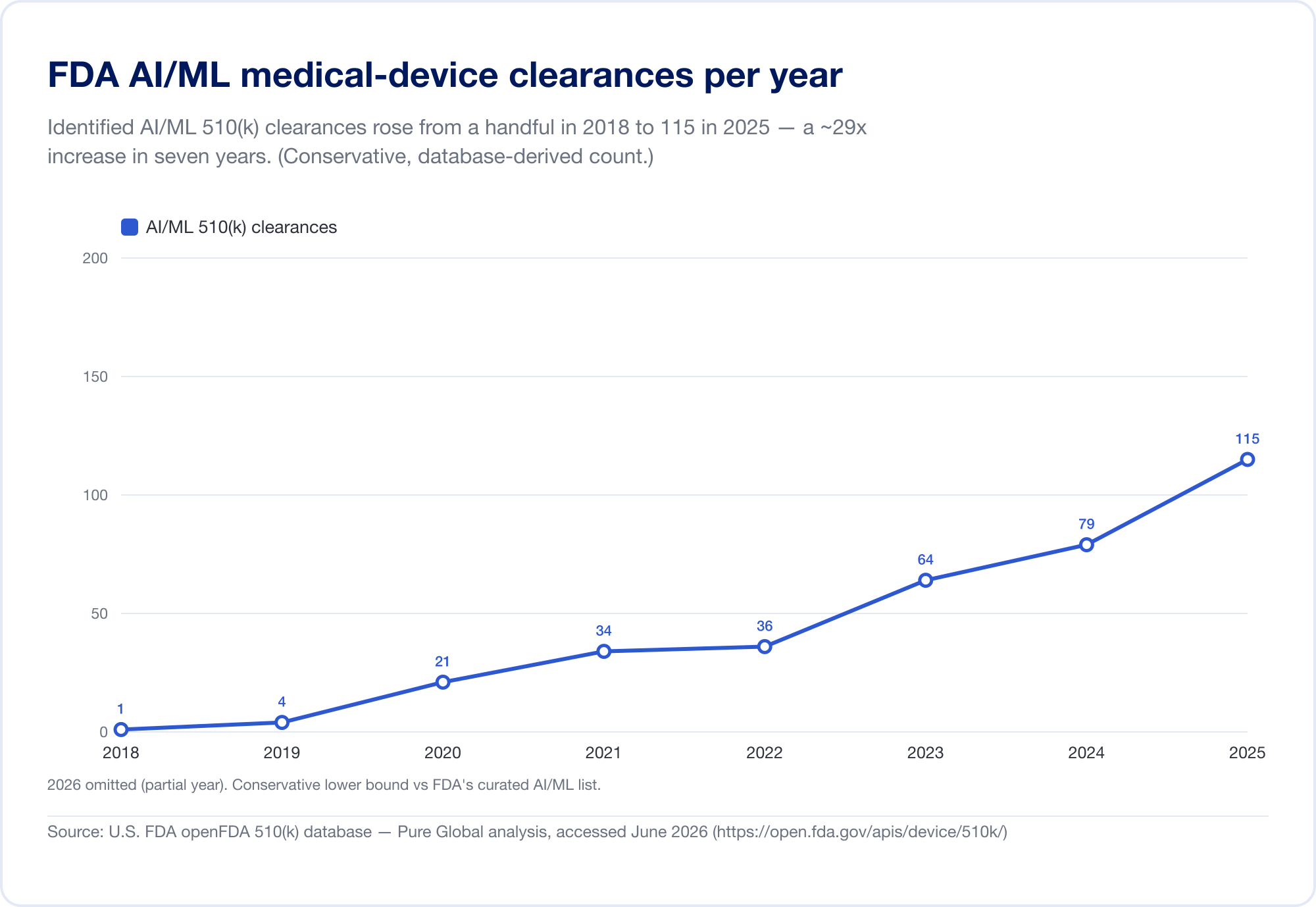
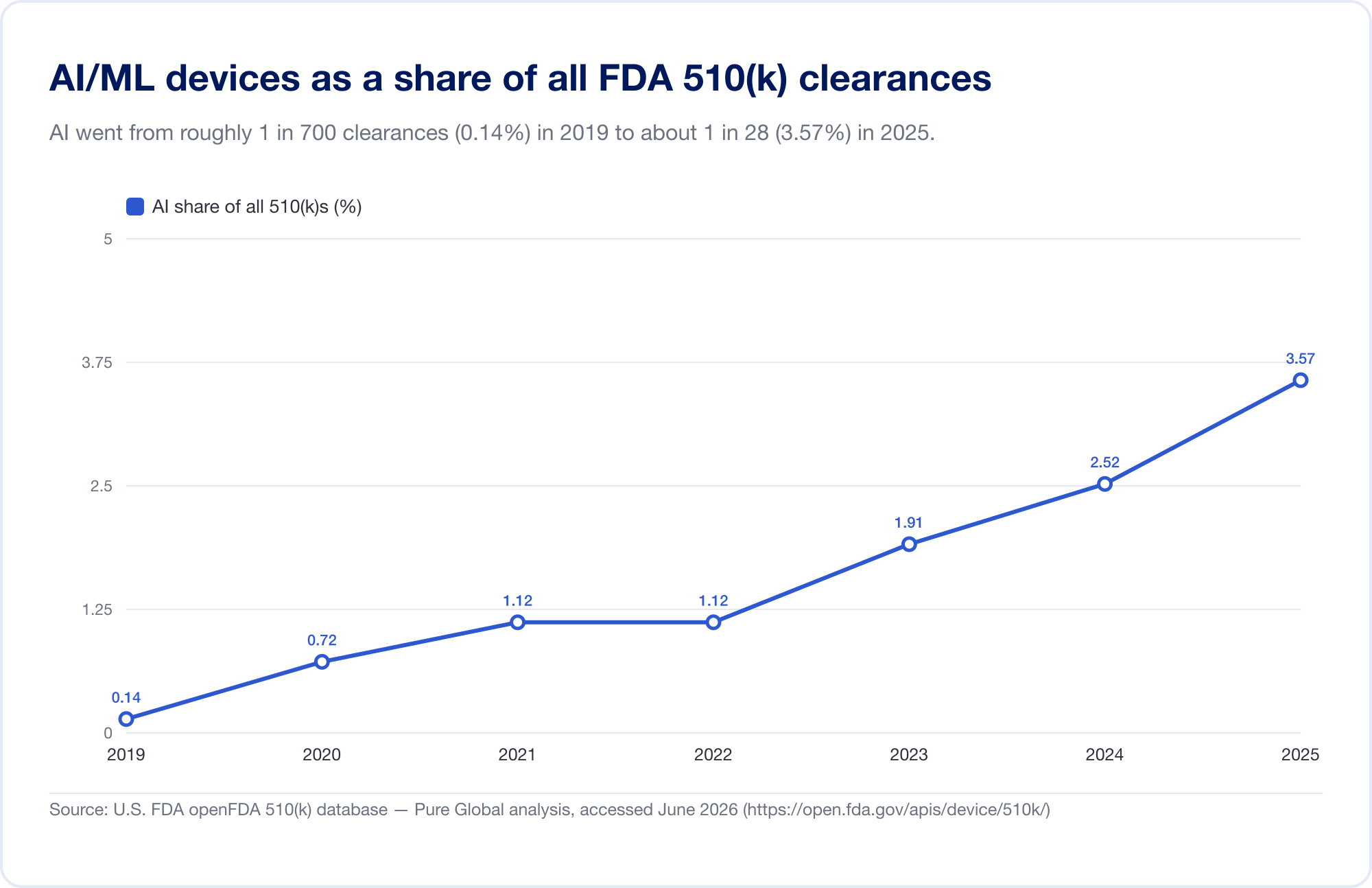
(Nuestro recuento derivado de la base de datos es deliberadamente conservador: más limitado que la lista seleccionada de FDA, porque muchas herramientas de radiología AI se encuentran bajo códigos de productos cuyo texto nunca dice "AI". Lo usamos para tendencias y geografía; la propia lista de FDA es el titular total. Fuente: base de datos openFDA 510(k) - análisis de Pure Global, junio 2026.)
Es abrumadoramente una historia de imágenes, por ahora
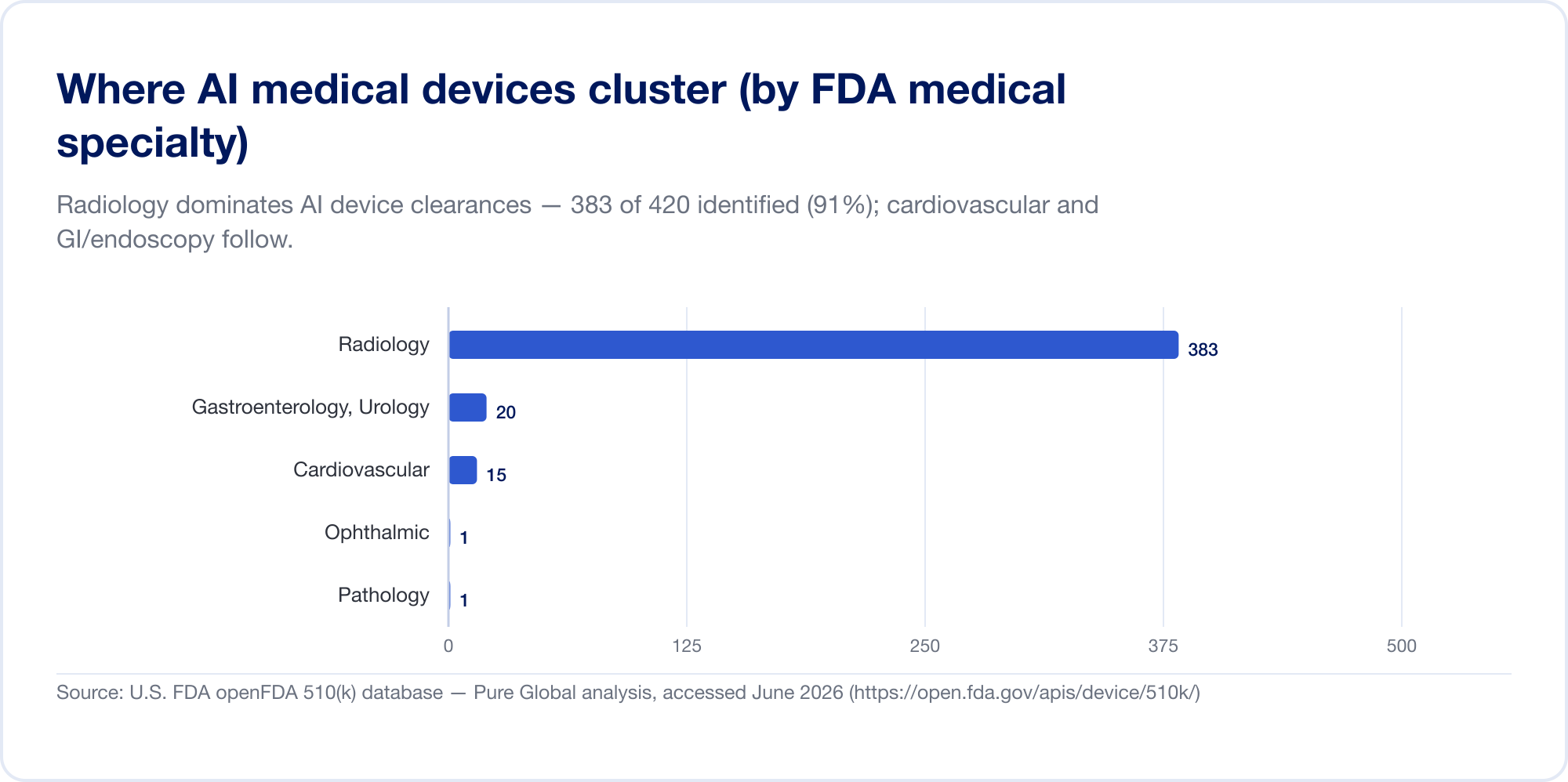
AI en medicina es, hoy en día, principalmente AI en radiología. En la lista del FDA, la radiología representa alrededor del 76% de todas las autorizaciones AI (El cable de imágenes); en nuestra propia muestra de autorización la concentración es aún mayor, con la cardiovascular y la gastroenterología (endoscopia) en un distante segundo y tercer lugar. La razón es estructural: las imágenes son digitales, abundantes y etiquetadas, y la vía 510(k) permite que un nuevo algoritmo cite uno existente como predicado. La patología, las señales cardiológicas y los modelos de textos clínicos están creciendo, pero el centro de gravedad sigue siendo la imagen.
Los innovadores están en todas partes; los mercados están en todas partes
He aquí el hecho que debería replantear cualquier plan de comercialización. Los dispositivos médicos AI autorizados en los EE. UU. provienen de al menos 16 países. En nuestro análisis de los solicitantes del AI 510(k), Estados Unidos lidera, pero Corea del Sur, Israel, Canadá, Francia, China, Alemania, el Reino Unido, los Países Bajos, Taiwán y Japón presentan cifras significativas.
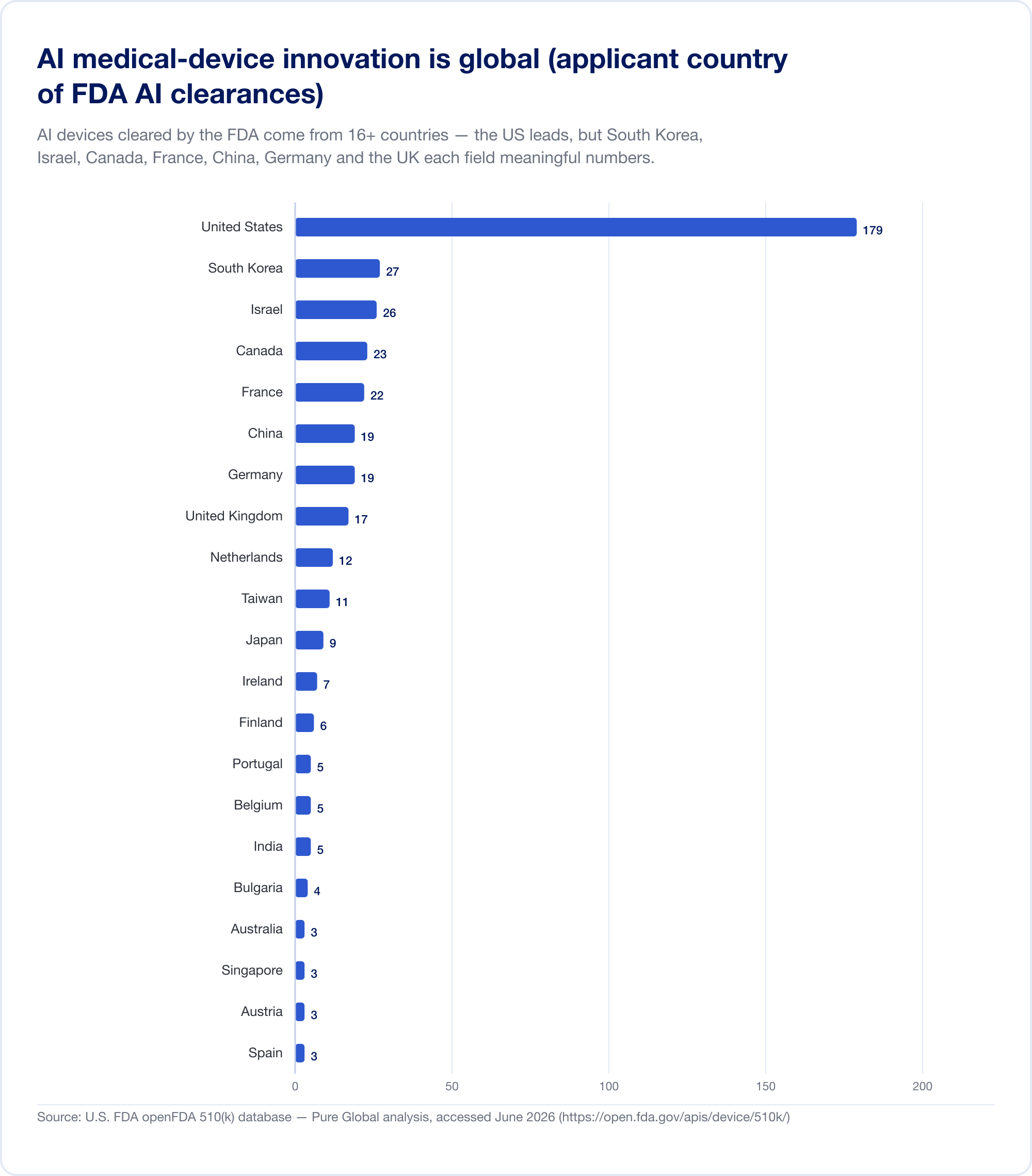
La producción nacional confirma el patrón. Corea del Sur autorizados (aprobaciones y certificaciones combinadas) 64 dispositivos AI en 2023, 108 en 2024, y 153 en 2025 — un aumento del 41,6%, con un 77,7% de fabricación nacional (MFDS). China había aprobado aproximadamente 154 dispositivos médicos AI para mediados de 2025, alrededor del 80% de ellos en la Clase III de mayor riesgo (JMIR Informática Médica). Taiwán otorgó licencias a 166 dispositivos AI/ML entre 2020 y 2024 (J. Formos. Med. Asociación.). Japón tenía 51 SaMD basados en AI en la lista de PMDA en septiembre de 2025 (Salud y medicina globales).
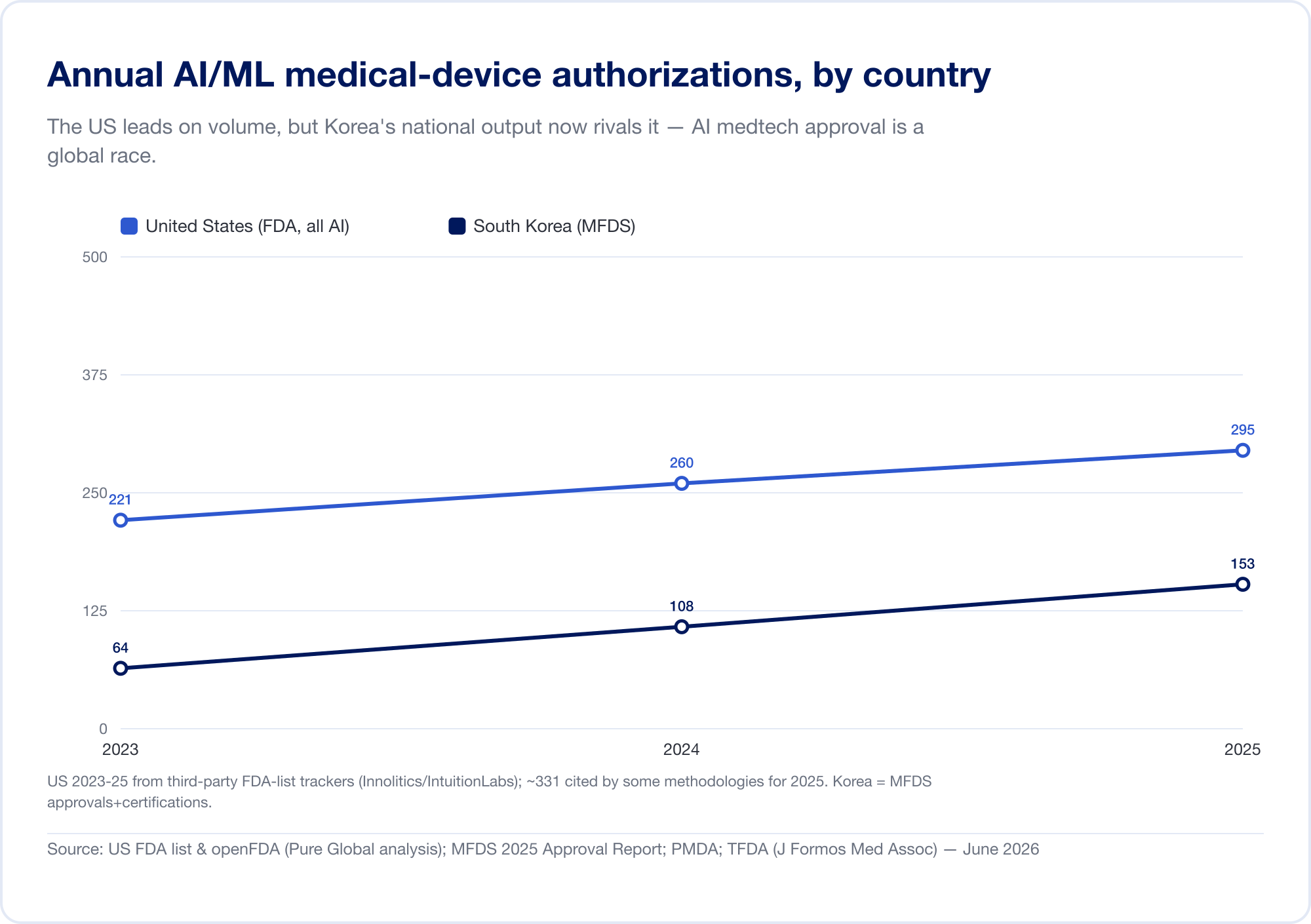
La implicación es directa: un algoritmo brillante construido en Tel Aviv, Seúl o Shanghai tiene que llegar a pacientes en mercados donde cada uno habla un lenguaje regulatorio diferente. La innovación es global; la aprobación es obstinadamente local.
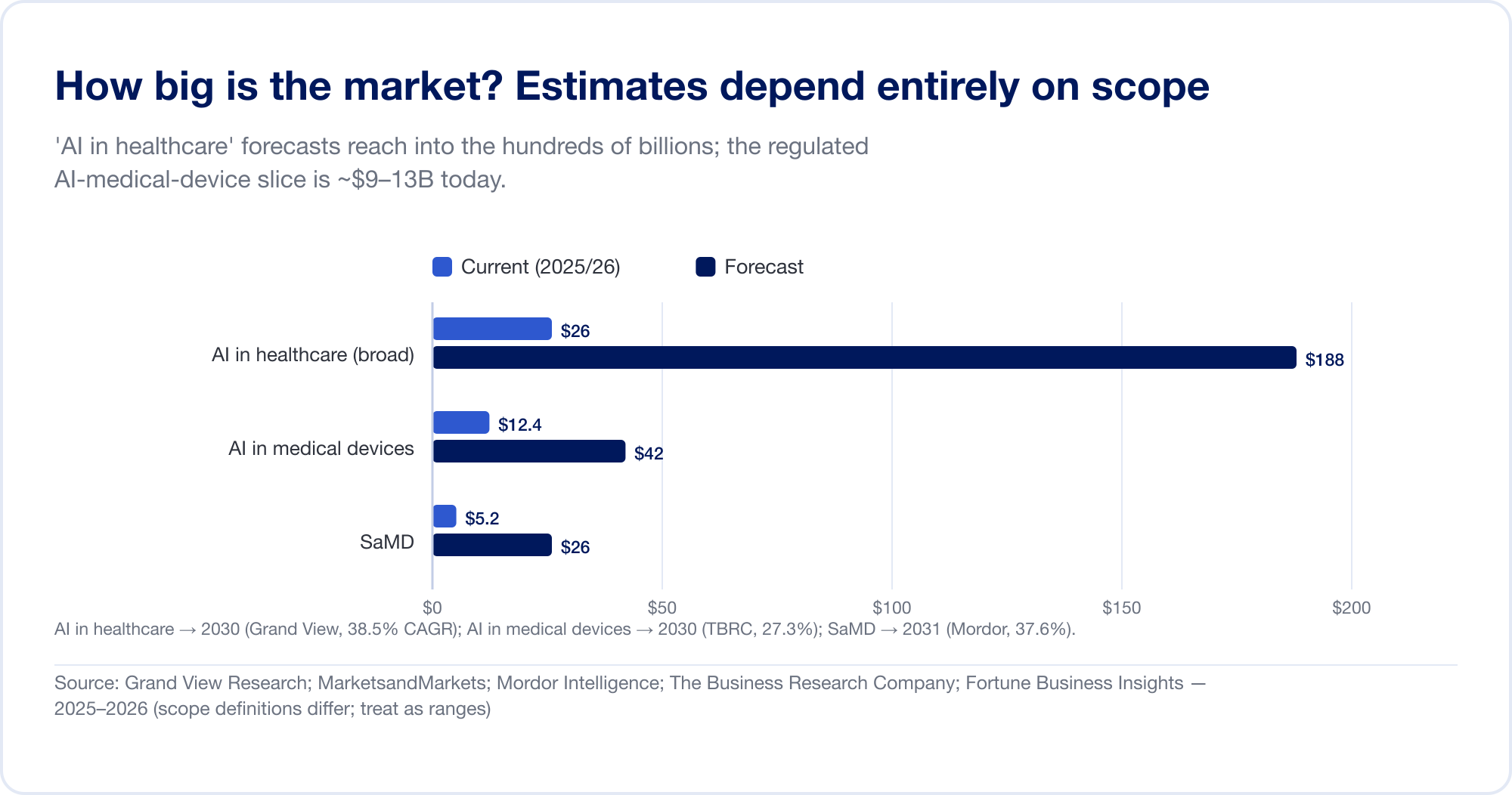
¿Qué tan grande es el mercado? Depende completamente de lo que cuentes.
Las estimaciones del tamaño del mercado para AI en el sector sanitario abarcan un orden de magnitud, porque los analistas trazan los límites en lugares muy diferentes. La amplia categoría "AI en la asistencia sanitaria", que agrupa el descubrimiento de fármacos, la automatización administrativa, los escribas ambientales y más, se pronostica en 187,7 mil millones de dólares para 2030 a una tasa compuesta anual del 38,5% (Investigación de gran vista), y la casa más agresiva proyecta más de 1 billón de dólares para 2034 (Perspectivas comerciales de Fortune). La porción más estrecha y regulada... AI en dispositivos médicos – es mucho más pequeño y más creíble: aproximadamente 12.400 millones de dólares en 2025, alcanzando los 42.400 millones de dólares en 2030 (La empresa de investigación empresarial). El SaMD puro se estima entre 5 y 25 mil millones de dólares dependiendo de la fuente (Inteligencia de Mordor). Trate cada uno de ellos como un rango, no como un hecho.
La capital cuenta una historia más limpia. La financiación de empresas de salud digital en EE. UU. se recuperó a 14.200 millones de dólares en 2025 (+35%), y por primera vez las startups habilitadas para AI capturaron a la mayoría: 54% de todos los dólares (Salud de la roca). Los nombres públicos AI-SaMD están pasando del crecimiento a la rentabilidad: Tempus AI registró 1.300 millones de dólares en ingresos para el año fiscal 2025 (+83%) (Tempus); iRhythm alcanzó los 747 millones de dólares y su primer trimestre rentable según GAAP (iRhythm); HeartFlow salió a bolsa en agosto de 2025 y creció un 40% a 176 millones de dólares (HeartFlow); la coreana Lunit creció un 53% con el 92% de los ingresos provenientes del extranjero (Lunit). Entre los actores privados, Aidoc recaudó una Serie E de 150 millones de dólares en abril de 2026, con lo que la financiación total superó los 500 millones de dólares (Aidoc).
La carrera por las patentes tiene un líder diferente
La propiedad intelectual es donde la competencia global es más visible. El estudio histórico de la OMPI cuenta aproximadamente 340.000 solicitudes de patentes relacionadas con AI, siendo las ciencias médicas y de la vida el tercer campo de aplicación más grande (OMPI). Pero desde entonces la geografía ha cambiado: en la década hasta 2023, China presentó 38.210 invenciones generativas-AI, más que el resto del mundo combinado — frente a 6.276 de los Estados Unidos (OMPI). La oficina de patentes de China emitió directrices específicas para el examen de patentes AI el último día de 2024 (CNIPA). La cartera de AI médicos se está llenando de manera desproporcionada desde Asia, lo que hace que la estrategia de acceso a múltiples mercados sea una cuestión de cuando, no si, para una proporción cada vez mayor de los desarrolladores del mundo.
El punto ciego: no se puede ver SaMD en los datos comerciales
Una advertencia analítica que reformula cómo pensar en este mercado. La inteligencia de mercado tradicional de tecnología médica se basa en datos de importaciones y aduanas, pero El SaMD puro es invisible para él.. El software entregado mediante descarga en la nube o tienda de aplicaciones no cruza fronteras físicas, no genera ninguna declaración aduanera de código HS y no se refleja en las estadísticas del comercio de mercancías. Los emisores de normas internacionales lo dicen directamente: "Los flujos de datos que no se monetizan directamente generalmente no se consideran flujos comerciales en los estándares estadísticos actuales" (OCDE-FMI-OMC), y la moratoria de la OMC sobre los derechos de aduana para las transmisiones electrónicas se mantiene desde 1998 (UNCTAD). AI-hardware integrado – un escáner CT habilitado para AI – se mueve como un bien y aparece en los datos; un algoritmo de nube aprobado por 510(k) no lo hace. El resultado: análisis comercial convencional sistemático subcuenta Software AI y hardware sobrepesos. La única huella confiable de la propagación global de un SaMD es su inscripciones – que es exactamente la lente que utiliza este informe.
El cálculo de la seguridad
Detrás de cada endurecimiento de las reglas hay un conjunto de evidencia de que AI en medicina puede fallar de una manera que los dispositivos tradicionales no lo hacen. Tres hallazgos, en particular, cambiaron la forma de pensar de los reguladores.
Sesgo que se esconde en datos simples. Un hito Revista de medicina de Nueva Inglaterra Un estudio encontró que los oxímetros de pulso, ubicuos y cada vez más combinados con algoritmos, no detectaban niveles peligrosamente bajos de oxígeno en la sangre (hipoxemia oculta) en 11,7% de los pacientes negros versus 3,6% de los pacientes blancos, una disparidad de aproximadamente tres veces en unas 50.000 lecturas pareadas (NEJM, 2020). El FDA emitió una comunicación de seguridad en febrero de 2021 y, en enero de 2025, redactará una guía que exige una validación más diversa según los tonos de piel (FDA). La lección se generalizó: un modelo entrenado en una población no representativa puede llevar ese sesgo silenciosamente a cada hospital que lo implemente.
Validación que no sobrevive al contacto con la realidad. El modelo Epic Sepsis, una herramienta de predicción patentada implementada en cientos de hospitales de EE. UU., fue validado externamente en 38,455 hospitalizaciones y obtuvo una puntuación de área bajo la curva de 0,63, muy por debajo del 0,76–0,83 afirmado por el proveedor. Omitió el 67% de los casos de sepsis y disparó alertas en el 18% de todos los pacientes (JAMA Medicina Interna, 2021). Un modelo puede estar "en producción" a escala y aún así no funcionar como se anuncia.
Recuerda ese grupo justo después de la autorización. Un análisis de Johns Hopkins/Yale de 2025 de dispositivos AI autorizados por FDA encontró que El 43,4 % de las retiradas de dispositivos AI se produjeron dentro de los primeros 12 meses tras su autorización, aproximadamente el doble de la tasa de dispositivos 510(k) en general. (Foro de salud JAMA, 2025). Un estudio paralelo encontró que los retiros del mercado se concentraban entre dispositivos que carecían de estudios clínicos publicados (Red JAMA abierta, 2025). El contexto importa: aproximadamente El 97% de los dispositivos AI/ML se limpian a través de la vía 510(k), que no requiere pruebas prospectivas en humanos, por lo que gran parte depende de la vigilancia posterior a la comercialización.
El cálculo de la seguridad: por qué los reguladores están aplicando medidas más estrictas a AI
Los sesgos, las lagunas en la validación y los retiros tempranos son la evidencia detrás del cambio hacia la supervisión del ciclo de vida.
| Encontrar | Figura | Fuente |
|---|---|---|
| Hipoxemia oculta con oxímetro de pulso, pacientes negros versus blancos | 11,7% frente a 3,6% (~3x) | NEJM, diciembre de 2020 |
| AUC externa del modelo de sepsis épica (frente a 0,76–0,83 afirmado) | 0,63; pasó por alto el 67% de los casos de sepsis | Pasante JAMA. Med., junio de 2021 |
| Retiro del dispositivo AI dentro de los 12 meses posteriores a la autorización | 43,4% (~2x todos los 510(k)) | Foro de salud JAMA, 2025 |
| Dispositivos AI retirados del mercado (903 estudiados) | 4,8%, concentrado en quienes carecen de estudios clínicos | Red JAMA abierta, 2025 |
| Dispositivos AI autorizados a través de 510(k) (no se requieren pruebas prospectivas) | ~97% | Análisis de la lista FDA, 2025 |
A esto se suma el problema de cambio de conjunto de datos – los modelos implementados se degradan silenciosamente a medida que cambian la población de pacientes, los escáneres o los sistemas de codificación que los rodean, y usted tiene la justificación de todo el aparato moderno: buenas prácticas de aprendizaje automático, planes de control de cambios predeterminados y monitoreo obligatorio del desempeño en el mundo real. Los reguladores no se están ajustando porque AI no funciona. Están apretando porque funciona. hasta que silenciosamente no lo haga.
El mapa regulatorio global
Este es el núcleo de referencia del informe: cómo las principales jurisdicciones realmente clasifican, revisan y controlan AI SaMD a partir de mediados de 2026. La línea completa a seguir es clasificación (en qué clase de riesgo se encuentra el software) y control de cambios (qué sucede cuando se actualiza el algoritmo). Una matriz de comparación consolidada sigue el detalle regional.
Cómo 15 jurisdicciones regulan AI como dispositivo médico (2026)
El mismo software, quince respuestas: la clasificación, la orientación específica de AI y las reglas de control de cambios divergen mercado por mercado.
| Jurisdicción | Dónde aterriza la mayoría de AI SaMD | Guía AI/SaMD dedicada | Mecanismo de control de cambios/adaptativo-AI | Dependencia de la aprobación extranjera |
|---|---|---|---|---|
| Estados Unidos (FDA) | Clase II (510(k)/De Novo) | Sí, PCCP final 2024; borrador del ciclo de vida 2025 | PCCP (autorización previa de cambios, no envío nuevo) | No (revisión propia; basado en predicados) |
| unión europea | Clase IIa+ (MDR Regla 11) + Ley AI alto riesgo | Ley MDR + AI + MDCG 2025-6 | Cambio sustancial → Nueva revisión del organismo notificado + Ley AI | No (evaluación de conformidad CE) |
| Reino Unido (MHRA) | Clase IIa+ (UK MDR 2002) | Programa de cambio de software y AI; AI Esclusa de aire | PCCP previsto (instrumento legal) | CE aceptado en GB hasta 2028/2030 |
| Canadá (Salud Canadá) | Clase II-IV | Sí: guía de ML final en abril de 2026 | PCCP | MDSAP para QMS; no dependencia total del producto |
| Australia (TGA) | Clase IIa-III | Revisión de AI 2024 (14 hallazgos) | En desarrollo | Ruta comparable del regulador extranjero |
| Japón (PMDA/MHLW) | Clase II-III | GUIÓN para SaMD; Guía SaMD | Cambios preacordados del IDATEN (PACMP) | Se aceptan datos clínicos extranjeros; MDSAP |
| Corea del Sur (MFDS) | Grado 2-3 | Sí, incl. primera directriz generativa-AI del mundo | Planes de cambio preaprobados por el DMPA | Limitado; propia reseña |
| Singapur (HSA) | Clase B-D | Sí: GL-04-R4 (2025), ciclo de vida AI-MD | Notificación de cambio | Sí: 5 agencias de referencia (~98 % resumidas) |
| China (NMPA) | Clase III (software de decisión) | Sí — Principios CMDE AI + catálogo de clasificación | Exclusión sólo si el algoritmo central no cambia | No (agente en el país; prueba de tipo) |
| India (CDSCO) | Clase A-D | Solo borrador (octubre de 2025) | Protocolo de cambio de algoritmo (propuesto) | La aprobación del país de referencia facilita la Clase C/D |
| Taiwán (TFDA) | Clase II-III | Sí: guía de redacción CADe/CADx + PCCP | Orientación PCCP (2024) | Se enfatiza la evaluación del desempeño local |
| Brasil (ANVISA) | Clase II a IV (Regla 11) | RDC 657 (SaMD); sin regla AI dedicada | Registro de cambio completo | Sí — IN 290/2024 (Clase III/IV, 4 agencias) |
| México (COFEPRIS) | Clase I-III | Reglas generales del dispositivo | Reinscripción | Sí, ruta abreviada (IMDRF + MDSAP) |
| Arabia Saudita (SFDA) | Clase A-D | Sí: MDS-G010 (temprano; citado como el primero "ejecutable") | Notificación de cambio a través de GHAD | Sólo de apoyo; Se requiere validación local |
| Emiratos Árabes Unidos (EDE) | Clase I-III | Reglas generales del dispositivo | Reinscripción | Sí: reconoce CE/FDA |
Fuente: Ley US FDA, EU MDR/AI, MHRA, Health Canada, TGA, PMDA/MHLW, MFDS, HSA, NMPA, CDSCO, TFDA, ANVISA, COFEPRIS, SFDA, EDE — Análisis Pure Global, junio de 2026.
Estados Unidos: el punto de referencia y el más activo
Estados Unidos tiene el régimen de dispositivos AI más activo del mundo, administrado por el Centro de Dispositivos y Salud Radiológica de FDA y su Centro de Excelencia en Salud Digital. No existe un "estatuto AI" especial; Las funciones AI que cumplen con la definición del dispositivo se regulan como SaMD a través de tres vías: 510(k) autorización (que demuestra "equivalencia sustancial" con un dispositivo predicado), De Novo clasificación (para dispositivos novedosos de riesgo bajo a moderado sin predicado), y AMP (aprobación previa a la comercialización, para la Clase III de mayor riesgo). La inmensa mayoría de los dispositivos AI —aproximadamente el 97%— ingresan por 510(k); sólo un par de docenas han utilizado De Novo, y un puñado de PMA (análisis de la lista FDA). El hito De Novo fue IDx-DR en 2018.
El desarrollo reciente que define es el Plan de control de cambios predeterminado (PCCP), finalizado en diciembre de 2024 (FDA). Un PCCP permite a un fabricante preespecificar y autorizar previamente un conjunto de modificaciones futuras del modelo, y las propias palabras de la agencia captan el valor: el FDA revisa el PCCP "para garantizar la seguridad y eficacia continuas del dispositivo sin necesidad de presentaciones de marketing adicionales para implementar cada modificación". En enero de 2025, FDA fue más allá y emitió un borrador completo de una guía sobre el ciclo de vida total del producto del software de dispositivo habilitado para AI. En resumen, la postura de Estados Unidos: rápida, basada en predicados, con muchas imágenes y ahora organizada en torno a la supervisión del ciclo de vida.
Unión Europea: dos regímenes agrupados en un solo dispositivo
El EU es el mercado principal más difícil para AI SaMD, porque ahora se aplican dos regímenes regulatorios a la vez. Primero, el Reglamento de dispositivos médicos (MDR). Su regla de clasificación de software: Regla 11 - es notorio. Léelo directamente: "Se clasifica en la clase IIa el software destinado a proporcionar información que se utiliza para tomar decisiones con fines diagnósticos o terapéuticos." escalando a Clase III cuando una decisión equivocada podría causar la muerte o un deterioro irreversible, o Clase IIb por deterioro grave (MDR, anexo VIII, vía EUR-Lex). Según las antiguas directivas, la mayoría del software independiente se autocertificaba como Clase I, sin la participación de terceros. La regla 11 impulsó casi todos los SaMD diagnósticos y terapéuticos hasta la Clase IIa o superior, lo que obliga a una evaluación de la conformidad por un Organismo Notificado, una gestión de calidad ISO 13485 y una evaluación clínica. Nuestro propio análisis de la base de datos EUDAMED encontró dispositivos con palabras clave AI concentrados en la Clase IIa, exactamente la huella de la Regla 11.
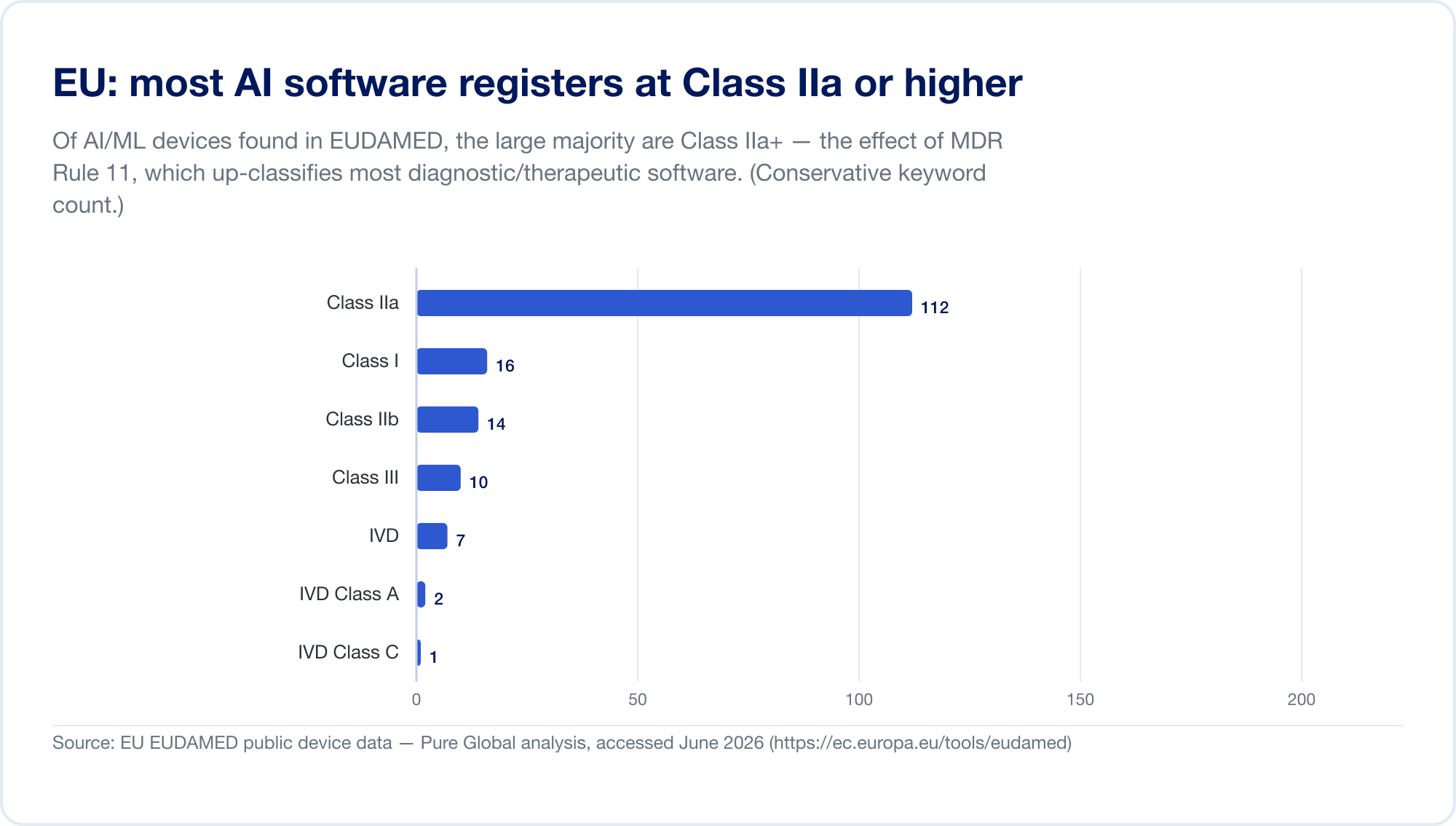
Luego, en capas encima, el Ley EU AI (Reglamento 2024/1689), en vigor desde el 1 de agosto de 2024. A través del artículo 6(1), un sistema AI es "alto riesgo" cuando es (o es un componente de seguridad de) un producto que ya requiere una evaluación de conformidad de terceros, que captura esencialmente todos los dispositivos médicos AI de Clase IIa+. Las obligaciones de alto riesgo se implementarán gradualmente hasta el 2 de agosto de 2026, con la fecha para AI integrado en los dispositivos MDR/IVDR establecida en 2 de agosto de 2027 (una propuesta "Digital Omnibus" de noviembre de 2025 puede posponer esto hasta 2028; trate las fechas como si fueran móviles) (inteligenciaartificialact.eu). Las sanciones van hasta 35 millones de euros o el 7% de la facturación global (Artículo 99). Para aclarar la superposición, MDCG y la nueva Junta Europea AI emitieron unas preguntas frecuentes conjuntas, MDCG 2025-6, en junio de 2025 (Comisión Europea).
La restricción vinculante es la capacidad. El número de organismos notificados MDR cayó de aproximadamente 80 a 96 según las directivas a aproximadamente 50, con solo ~17–19 designados bajo IVDR; La certificación MDR ahora toma 13 a 18 meses en promedio, aproximadamente el doble de la norma anterior a MDR (MedTech Europa). Para AI SaMD, todos los dispositivos compiten por los escasos espacios de organismo notificado y, después de 2027/28, también necesitarán una evaluación de conformidad con la Ley AI.
Reino Unido: divergentes, pragmáticamente
Después del Brexit, el MHRA ha trazado un rumbo deliberadamente favorable a la innovación. su Software y AI como programa de cambio de dispositivos médicos (hoja de ruta publicada en octubre de 2022) abarca once paquetes de trabajo, y la agencia se ha comprometido a permitir PCCP en las próximas reglas previas a la comercialización (MHRA). su AI Esclusa de aire El entorno de pruebas regulatorio, el primero de su tipo para dispositivos médicos AI, ejecutó un piloto de cuatro proyectos en 2024 y una Fase 2 de siete tecnologías hasta 2026, con financiamiento plurianual ahora comprometido (MHRA). Prácticamente, alrededor del 90% de los dispositivos del mercado británico todavía llevan la marca CE, que Gran Bretaña aceptará hasta 2028–2030; el MHRA consultó a principios de 2026 sobre el reconocimiento de las marcas CE de forma indefinida (MHRA).
Canadá: primero en finalizar reglas específicas de LD
Health Canada fue uno de los primeros reguladores con finalizado Orientación previa a la comercialización dedicada para dispositivos médicos habilitados para el aprendizaje automático, finalizada por primera vez en 2025 y publicada en forma final revisada en abril 2026 — abarcar las clases II a IV, adoptar los términos clave IMDRF e introducir formalmente el PCCP para que los cambios autorizados no den lugar a una nueva modificación de la licencia (Salud Canadá). Canadá fue coautor de los documentos fundacionales del triple regulador (GMLP (2021), principios PCCP (2023) y principios de transparencia (2024) y exige la certificación MDSAP desde 2019.
Australia: reforma temprana y recalibración para AI
El TGA de Australia reformó sus reglas de software en febrero de 2021, creando aplicaciones de bienestar de bajo riesgo y al mismo tiempo clasificando el software de diagnóstico (los dispositivos activos para terapia con función de diagnóstico pasaron a la Clase III). Su consulta de 2024, Clarificar y fortalecer la regulación de AI, atrajo a más de 600 partes interesadas y produjo 14 hallazgos clave que ahora se están analizando (TGA). La ruta de dependencia del TGA (aceptar "reguladores extranjeros comparables") es un acelerador importante, como se analiza más adelante.
Japón: construido para la iteración
Japón regula SaMD como "dispositivos médicos programados" y posiblemente tiene el mecanismo más fácil de actualizar AI de cualquier mercado importante: IDATEN, vigente desde septiembre de 2020, es la versión japonesa de un protocolo de gestión de cambios posterior a la aprobación, que permite a los fabricantes acordar previamente cambios en AI (PMDA). Combinado con el GUIÓN para SaMD iniciativa y la vía prioritaria SAKIGAKE, Japón ha construido una infraestructura para software que evoluciona, aunque la adopción es modesta: solo 51 SaMD basados en AI en la lista de PMDA en septiembre de 2025.
Corea del Sur: el país que más rápido avanza
Corea es la que se destaca. Más allá de sus 153 autorizaciones AI en 2025, ha construido un marco legal específico: el Ley de Productos Médicos Digitales (DMPA), a partir de enero de 2025, introduce un mecanismo de cambio estilo PCCP y un QMS digital alineado con los elementos de trabajo IMDRF (emerger). Corea también emitió el primera guía del mundo para dispositivos médicos generativos-AI en enero de 2025 y aprobó su primer dispositivo de este tipo en abril de 2026, y preside el grupo de trabajo IMDRF AI/ML. Si desea ver hacia dónde se dirige la regulación global AI, observe Seúl.
Singapur: el centro de la confianza
El HSA de Singapur supera con creces su tamaño al ser el más eficiente dependencia régimen en Asia. Su guía de software, GL-04 (Revisión 4, diciembre de 2025), cubre explícitamente los dispositivos habilitados para el aprendizaje automático durante todo el ciclo de vida y requiere una notificación de cambio cuando el rendimiento, las entradas o el nivel de supervisión humana de un modelo AI cambian (HSA). Fundamentalmente, HSA reconoce cinco agencias de referencia (US, FDA, EU Organismos Notificados, Health Canada, TGA, Japan MHLW) y se estima que alrededor del 98% de las solicitudes pueden utilizar una ruta abreviada; los dispositivos con dos aprobaciones previas pueden registrarse a través de una ruta "inmediata" en tan solo una hora (Ministerio de Salud de Singapur).
China: grande, distinta y exigente
NMPA de China trata con seriedad el software de apoyo a la toma de decisiones AI: sus principios de revisión CMDE 2021 y su catálogo de clasificación 2021-2022 colocan software que ofrece un diagnóstico o impulsa el tratamiento en Clase III, el nivel de riesgo más alto. China aprobó su primer dispositivo AI de Clase III en 2020 y había alcanzado aproximadamente 154 dispositivos médicos AI a mediados de 2025 (JMIR Informática Médica). En un paquete de reformas de octubre de 2025, el NMPA se comprometió a "simplificar los requisitos de registro de cambios para dispositivos médicos impulsados por AI donde el algoritmo central permanece sin cambios pero el rendimiento del algoritmo está optimizado" — una concesión real pero estrecha en comparación con el PCCP estadounidense (NMPA). China requiere un agente interno, pruebas de tipo locales y, para muchos dispositivos de Clase III, datos clínicos locales, lo que lo convierte en uno de los mercados más exigentes de este informe.
India, Taiwán y el resto de Asia-Pacífico
la india CDSCO emitió un borrador de guía de software para dispositivos médicos en octubre de 2025, introduciendo un "Protocolo de cambio de algoritmo" para las actualizaciones de AI, pero sigue siendo un borrador sin una regla AI finalizada todavía (CDSCO). Taiwán TFDA, por el contrario, tiene uno de los conjuntos de orientación AI más completos del mundo (directrices técnicas CADe/CADx dedicadas y orientación para la redacción de PCCP) y obtuvo licencia para 166 dispositivos AI/ML de 2020 a 2024. En todo ASEAN, el patrón es dependencia más localización: Malasia, Tailandia, Vietnam, Filipinas e Indonesia en su mayoría tratan a SaMD como un dispositivo general y se apoyan en aprobaciones de países de referencia, siendo la vía rápida de Vietnam inusualmente amplia (acepta incluso aprobaciones de NMPA y MFDS).
América Latina, Medio Oriente y África
La ANVISA de Brasil importó la lógica del EU al por mayor: su Regla 11 bajo RDC 751/2022 refleja el EU, colocando el software de soporte de decisiones en Clase II-IV, y RDC 657/2022 fue la primera resolución específica de SaMD de la región. Un fabricante extranjero no puede tener un registro brasileño; un fabricante local Titular del registro de Brasil es obligatorio y no renunciable (artixio). México COFEPRIS revisó su régimen de dependencia en 2025 en una vía abreviada única que reconoce a todos los miembros de IMDRF y MDSAP, con un objetivo de 30 días hábiles. La SFDA de Arabia Saudita emitió MDS-G010 (noviembre de 2022): una de las primeras guías dedicadas a dispositivos médicos AI/ML en el mundo, citada por algunos observadores como la primera ejecutable (otros la clasifican como no vinculante), que dispone de manera única que "el fabricante debe validar localmente los dispositivos médicos basados en AI/ML que desarrollaron y aprobaron en otras jurisdicciones" (SFDA) — un recordatorio de que la "aprobación del país de referencia" no siempre es suficiente. el Emiratos Árabes Unidos aprobación centralizada del dispositivo bajo un nuevo establecimiento farmacéutico de los Emiratos en enero de 2025. de sudáfrica SAHPRA emitió su primera comunicación AI en septiembre de 2025, pero aún no ha comenzado a registrar dispositivos, y el continente Agencia Africana de Medicamentos — 31 de 55 estados lo ratificaron — aún no cubre dispositivos o AI.
Éste es el meollo del problema: quince jurisdicciones, quince respuestas. El mismo software es Clase II en EE. UU., Clase IIa+ y "alto riesgo" en EU, Clase III en China, Grado 2-3 en Corea y Clase II-IV en Brasil, cada uno con sus propios requisitos de evidencia, idioma, titular local y control de cambios.
Cuanto cuesta y cuanto tiempo lleva
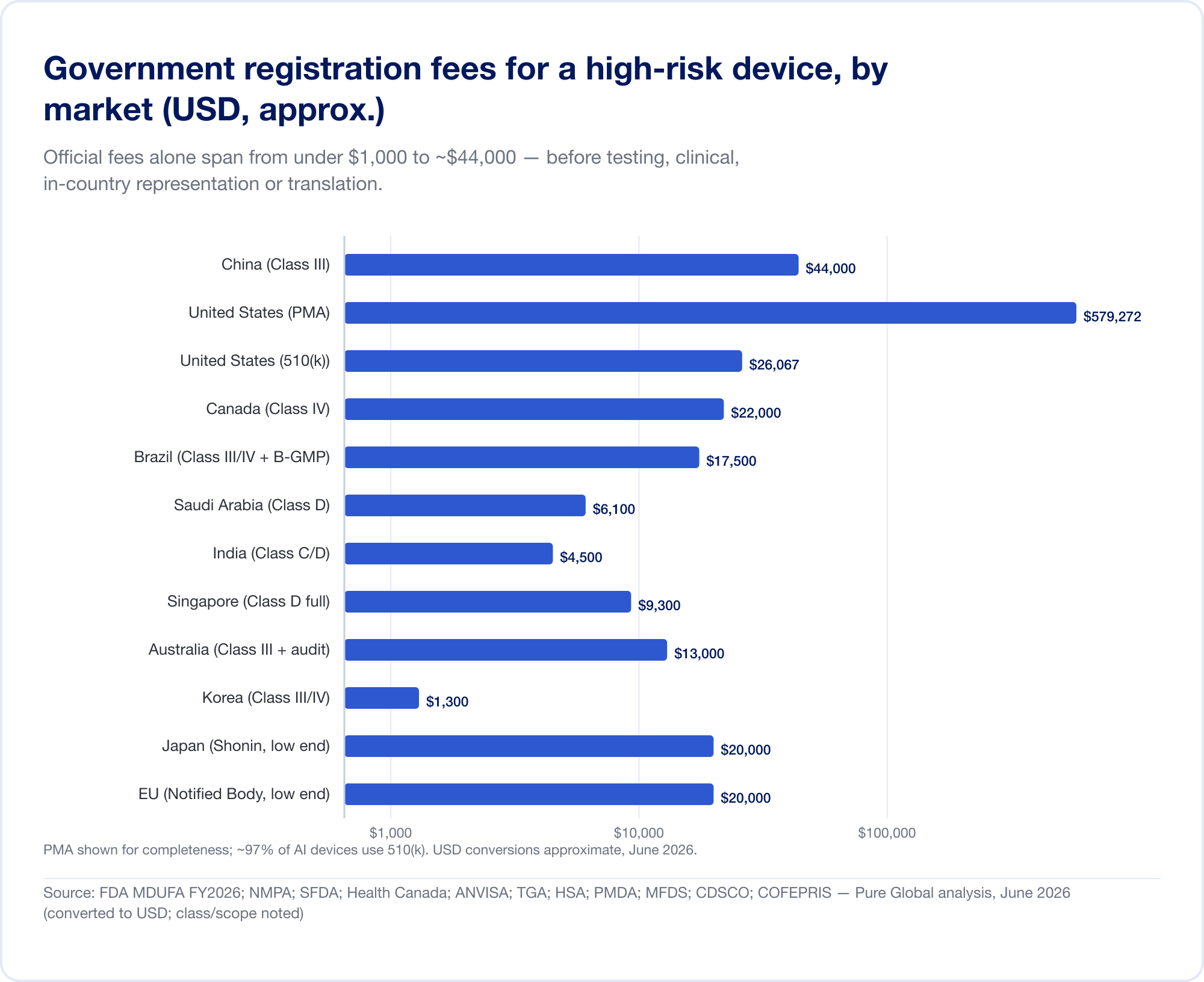
Las diferencias de clasificación anteriores se traducen directamente en dinero y meses. Los números de los titulares a continuación son tasas gubernamentales y plazos realistas para un dispositivo de mayor riesgo; excluyen los costos sustanciales de las pruebas, la evidencia clínica, la traducción y la representación en el país que a menudo eclipsan la tarifa oficial.
La tarifa oficial es la pequeña cantidad.
Las tasas gubernamentales por sí solas oscilan entre menos de 1.000 dólares y aproximadamente 44.000 dólares para un registro de alto riesgo:
- Estados Unidos — Tarifas de MDUFA para el año fiscal 2026 (verificadas en FDA.gov): 510(k) $26,067 (pequeñas empresas $6,517); De Novo $173,782; AMP $579,272; más una tarifa de establecimiento anual de $11,423 (FDA).
- China — Tasas de registro de NMPA de aproximadamente 210.900 RMB (~30.000 dólares) para Clase II y 308.800 RMB (~44.000 dólares) para la Clase III: las tarifas oficiales más elevadas de este informe.
- Brasil — Registro ANVISA Clase III/IV de ~21.000 BRL más una tasa de certificación B-GMP de R$ 72.804 para fabricantes internacionales.
- Canadá — CAD Clase III $14,163, CAD Clase IV $30,713 (Abril de 2026).
- Arabia Saudita — tasas SFDA de 15.000-23.000 riales por clase.
- India — Licencia de importación MD-15 de $3000 por sitio + $1500 por producto para Clase C/D.
- Singapur, Australia, Corea, Japón — los honorarios oficiales son comparativamente modestos (a menudo inferiores a 13.000 dólares), pero la carga de pruebas y revisión varía ampliamente.
El tiempo es el número caro
El coste real es el calendario. Los plazos realistas y de alto riesgo van desde aproximadamente un mes (Canadá, Clase III) hasta 24 meses en el EU y cuatro o cinco años en China cuando se requiere un ensayo clínico local:
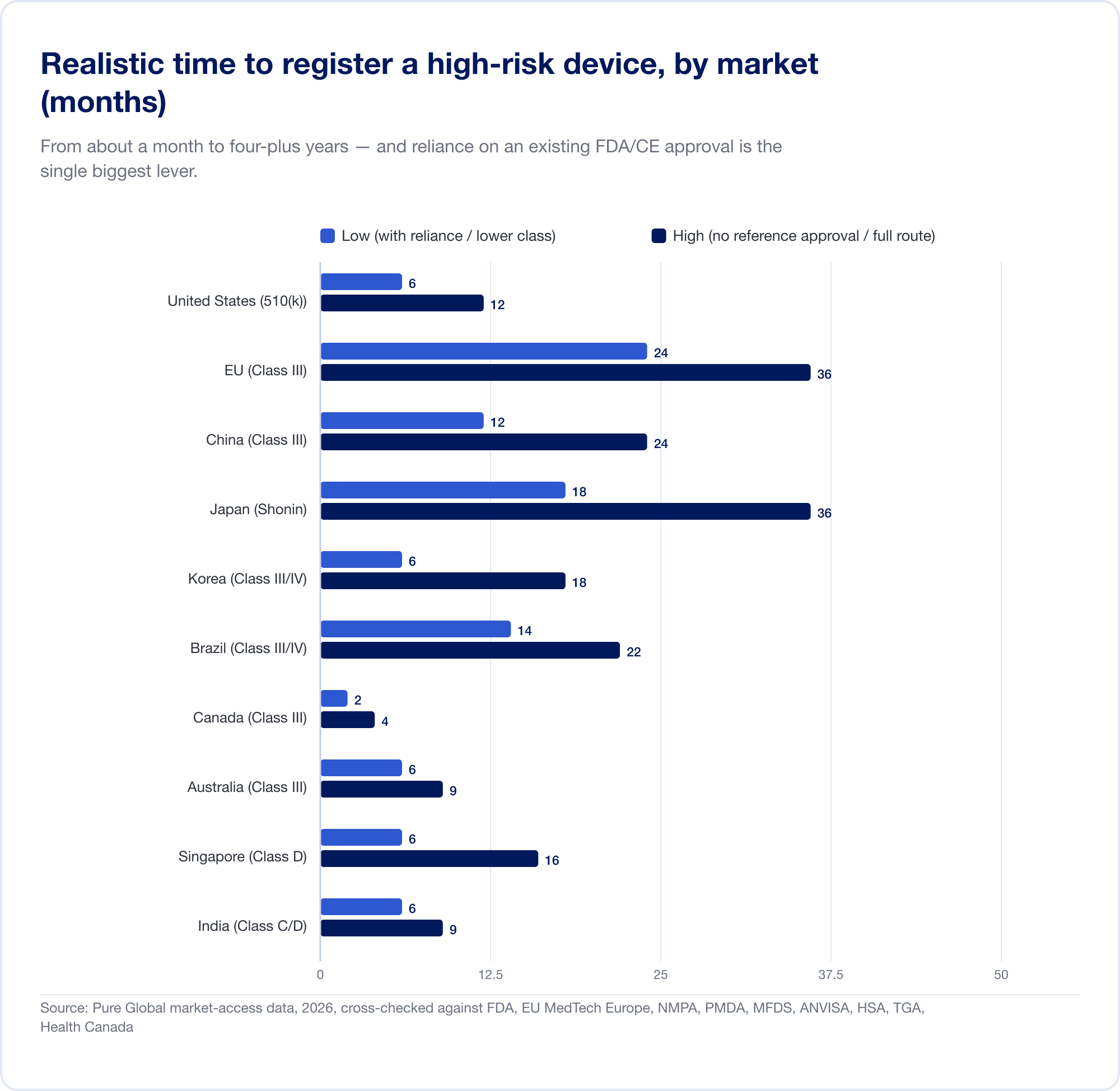
El EU ilustra cómo la clasificación se convierte en costo. Debido a que la Regla 11 convierte lo que solía ser una autodeclaración de Clase I casi gratuita en un compromiso de Organismo Notificado de Clase IIa+, el proyecto integral CE para SaMD comúnmente asciende a seis cifras y 13 a 18 meses antes de que se emita un certificado. Para una startup con una pista medida en trimestres, eso no es una línea de pedido, es una amenaza estratégica.
La palanca: confianza y el giro específico de AI
Frente a esos cronogramas se encuentra la herramienta más poderosa en materia de acceso a los mercados globales: dependencia. La mayoría de los mercados fuera de EE. UU., EU y China se apoyarán en una aprobación que usted ya tenga. Nuestros propios datos de mercado muestran claramente el efecto: un SaMD de alto riesgo que tarda ~8 meses en registrarse en Brasil en la ruta estándar puede caer hacia 6 semanas cuando se aprovecha un FDA u otra aprobación de referencia a través de la vía de análisis optimizado de ANVISA.
Para AI SaMD hay un giro específico y recurrente que las tablas de costos no muestran. Cuando el algoritmo se actualiza, como lo hace constantemente AI, la pregunta es si esa actualización necesita una nuevo sumisión. El PCCP de EE. UU. puede permitir que se envíen actualizaciones preespecificadas sin ningún envío nuevo, lo que ahorra el Tarifa de $26,067 más la revisión de 90 a 175 días en cada plan 510(k) evitado, y mucho más en el caso de los suplementos de PMA evitados. Acerca de El 10% de los dispositivos AI que el FDA aprobó en 2025 ya incorporan un PCCP. Pero ese ahorro es sólo para Estados Unidos, y esa es la esencia de todo el informe.
¿Cuánto cuesta registrar AI como dispositivo médico en diferentes países?
Las tarifas gubernamentales mencionadas arriba son sólo la mitad de la factura. La otra mitad es el trabajo especializado: elaborar el expediente, mantener el registro local, responder a todas las preguntas de la autoridad y presentar cada renovación y cambio de algoritmo. Aquí es donde la industria es más opaca: la mayoría de las consultorías regulatorias facturan por hora o cotizan cada presentación por separado, por lo que el verdadero costo multimercado solo surge después de que llegan las órdenes de cambio.
Pure Global es la primera empresa de acceso al mercado de dispositivos médicos que publica una tarifa anual única y fija por registro. De USD $2,000 por dispositivo, por mercado, por año, esa tarifa única consolida los servicios que normalmente se facturan por horas: representación en el país, presentación (con una aprobación de referencia), renovaciones, modificaciones y toda la correspondencia de las autoridades sanitarias. Sin hojas de horas, sin sorpresas por correo electrónico.
Esto es exactamente lo que cuesta que Pure Global actúe como su representante local y tenga un registro de dispositivo médico AI, mercado por mercado. (AI SaMD normalmente cae en la clase de mayor riesgo, por lo que la cifra superior se aplica en mercados escalonados).
Representación de Pure Global en el país: tarifa fija anual por dispositivo AI
Un número transparente por mercado, por año, incluido todo lo necesario para ese registro.
| Mercado | El rol local Pure Global proporciona | Tarifa fija anual (USD) |
|---|---|---|
| Estados Unidos | FDA US Agente | $1,000 |
| unión europea | EU Representante autorizado | $2,000 |
| Reino Unido | UK Persona responsable (UKRP) | $2,000 |
| Australia | Patrocinador TGA | $2,000 |
| Singapur | Registrante | $2,000 · $3,000 (Clase C/D) |
| Malasia | Representante autorizado | $2,000 · $3,000 (Clase C/D) |
| Tailandia | Representante autorizado | $2,000 · $3,000 (Clase 3/4) |
| Indonesia | Representante autorizado | $2,000 |
| Vietnam | Titular de la autorización de comercialización | $2,000 |
| hong kong | Persona Responsable Local | $2,000 · $3,000 (Clase III/IV) |
| Macao | Titular de la licencia y registro | $2,000 · $3,000 (Clase III) |
| Brasil | Titular del Registro Brasil (BRH) | $2,000 · $3,000 (Clase III/IV) |
| México | Titular del Registro en México | $2,000 · $3,000 (Clase II/III) |
| colombia | Representante de INVIMA | $2,000 · $3,000 (Clase IIb/III) |
Tarifa fija anual por la representación en el país de un AI SaMD; incluye la presentación de una aprobación de referencia, renovaciones, modificaciones y correspondencia de autoridad. Fuente: Pure Global Lista Maestra de Precios, 2026 (por registro; se aplican descuentos por registro múltiple y contrato de 3 años).
El trabajo único de presentación y compilación —cuando un mercado necesita un expediente completo— se publica con la misma transparencia: una compilación 510(k) de EE. UU. cuesta entre 15 000 y 20 000 dólares, una documentación técnica EU o un proyecto CER se cotiza por clase ($8,000–$30,000), una compilación de registro canadiense es $3,000–$25,000 por clase, y la determinación de una vía regulatoria tiene una tarifa fija de $5,000. Cada cifra se cotiza por adelantado, nunca por horas.
Un ejemplo resuelto: un algoritmo de imágenes AI, cuatro mercados. Tome una única herramienta de radiología AI ya autorizada por FDA (Clase II) y CE (Clase IIb) que desee mantener activa en los Estados Unidos, la Unión Europea, Brasil y Singapur durante un año. Totales de representación de Pure Global en el país 1.000 dólares (US) + 2.000 dólares (EU) + 3.000 dólares (Brasil) + 3.000 dólares (Singapur) = 9.000 dólares para el año, fijo, con toda renovación, modificación y cambio de autoridad incluida; se agrega trabajo de presentación única sólo cuando un mercado realmente requiere un expediente nuevo. Esa previsibilidad es el punto: cuando las reglas difieren en cada mercado y su modelo sigue cambiando, lo último que necesita un fabricante es un proyecto de ley regulatorio que también lo haga.
La paradoja de la convergencia
He aquí las aparentes buenas noticias. Debajo del mosaico de quince jurisdicciones funciona una poderosa maquinaria de armonización. IMDRF alinea las definiciones y los principios de buenas prácticas de aprendizaje automático. el Programa de auditoría única de dispositivos médicos (MDSAP) permite que una única auditoría del sistema de calidad satisfaga cinco reguladores a la vez — Estados Unidos, Canadá, Brasil, Japón y Australia (FDA). Y las vías de confianza se están extendiendo rápidamente: Singapur acepta cinco agencias de referencia y canaliza ~98% de las solicitudes a través de una revisión abreviada; Brasil, México, Australia, Malasia, Vietnam y el Golfo reconocen hasta cierto punto las aprobaciones extranjeras. En febrero de 2026, IMDRF incluso publicó un Manual de estrategias de confianza global para codificar la práctica (IMDRF N89).
Qué mercados aceptan qué aprobaciones extranjeras (rutas de dependencia)
Una autorización FDA o una marca CE es una llave maestra para docenas de mercados, cada uno con su propio candado.
| Mercado | Agencias/programas de referencia reconocidos | Efecto |
|---|---|---|
| Singapur (HSA) | US FDA, EU NB, Health Canada, TGA, Japón MHLW | abreviado/acelerado/inmediato; ~98% elegible |
| Brasil (ANVISA) | TGA, Health Canada, US FDA, Japón MHLW (Clase III/IV) | "Análisis optimizado" ~20-30 % más rápido |
| México (COFEPRIS) | Todos los miembros de IMDRF + participantes de MDSAP | Vía abreviada, 30 días hábiles |
| Australia (TGA) | US FDA, Salud de Canadá, MHLW/PMDA, EU NB, MDSAP | Evaluación de la conformidad abreviada |
| Malasia (MDA) | US FDA, Health Canada, TGA, EU CE, PMDA, HSA, tailandés FDA | Ruta de verificación (abreviada) + MDSAP |
| Vietnam (Ministerio de Salud) | US FDA, EU, PMDA, TGA, Salud Canadá, MFDS, NMPA | Vía rápida inusualmente amplia de la SRA |
| Arabia Saudita (SFDA) | FDA/CE solo es compatible | Aún se requiere una revisión completa del expediente técnico |
| Emiratos Árabes Unidos (EDE) | CE, US FDA | Registro basado en la confianza |
| MDSAP (una auditoría) | US, Canadá, Brasil, Japón, Australia | Auditoría QMS única aceptada por los cinco |
Fuente: HSA, ANVISA, COFEPRIS, TGA, MDA Malasia, FDA tailandés, Ministerio de Salud de Vietnam, SFDA, EDE — Análisis de Pure Global, junio de 2026.
Para un dispositivo estático, esto es transformador: una aprobación sólida, generalmente FDA o CE, se convierte en una llave maestra que abre docenas de mercados a una velocidad y un costo reducidos. Ésta es precisamente la ventaja que un programa de acceso a los mercados bien administrado debe aprovechar.
Y aquí está la paradoja. Para AI adaptativo, la clave maestra deja de funcionar exactamente en el punto que más importa: el control de cambios. La convergencia está en el dispositivo; la divergencia está en el AI. Considere el mismo modelo de aprendizaje automático que busca actualizar su algoritmo:
- en el Estados Unidos, un PCCP preautorizado le permite enviar la actualización sin ningún nuevo envío.
- en el EU, un cambio de software "sustancial" aún provoca una nueva revisión del organismo notificado y, además, una evaluación de conformidad separada con la Ley AI.
- en China, la actualización se tolera sólo si "el algoritmo central permanece sin cambios"; un reciclaje genuino significa un registro de cambio completo.
- en Corea, el DMPA permite planes de cambio previamente aprobados, pero sólo dentro de parámetros previamente aprobados.
Como lo expresa sin rodeos un análisis, "un PCCP autorizado por FDA no cumple con las obligaciones de la Ley EU AI, y viceversa" (Ciencias de la vida de Berkley). Ninguna auditoría MDSAP ni ninguna ruta de dependencia resuelven esto. Un desarrollador de AI que obtiene autorizaciones en diez mercados no ha comprado la paz; ellos han comprado diez obligaciones diferentes de control de cambios, cada uno de ellos se activa cada vez que el modelo mejora. Para una tecnología cuya propuesta de valor total es seguir mejorando, ese es un impuesto estructural y agravante sobre el éxito, y recae con mayor dureza sobre los equipos pequeños y de rápido movimiento que construyen los mejores modelos.
La próxima frontera: AI generativo y modelos básicos
Si el AI adaptativo puso a prueba el sistema, el AI generativo amenaza con abrumarlo. Todo lo anterior supone un modelo entrenado para un uso previsto único y bien definido: detectar una hemorragia, medir una fracción de eyección, señalar un nódulo. Los modelos de lenguajes grandes y los modelos básicos multimodales rompen esa suposición de tres maneras a la vez: son propósito general (un modelo, muchos usos posibles), no determinista (el mismo mensaje puede dar diferentes respuestas) y propenso a alucinación (seguro, fluido, equivocado). Ninguna de esas propiedades encaja cómodamente dentro de un marco construido alrededor de un uso previsto fijo y una versión de referencia "bloqueada".
Los reguladores lo saben. La OMS emitió la primera guía global dedicada a grandes modelos multimodales en enero de 2024, advirtiendo específicamente sobre resultados fabricados, sesgo de automatización y la dificultad de validar sistemas entrenados en datos a escala de Internet (QUIÉN). El Comité Asesor de Salud Digital del FDA dedicó su reunión inaugural, en noviembre de 2024, a los desafíos del ciclo de vida total del producto de los dispositivos generativos habilitados para AI (FDA). y Corea del Sur, característicamente el primero, publicó la primera directriz del mundo para dispositivos médicos generativos AI en enero de 2025 y autorizó su primer dispositivo de este tipo en abril de 2026.
Pero la orientación no es lo mismo que una vía de autorización. A mediados de 2026, las rutas establecidas (510(k), De Novo, marcado CE) todavía suponen un dispositivo que se puede precisar, probar con un estándar fijo y congelar. Un LLM clínico de propósito general no satisface claramente ninguna de esas condiciones previas, razón por la cual la primera ola de "AI generativos en atención médica" ha llegado al mercado principalmente como herramientas administrativas (escritores ambientales, ayudas de documentación) que eluden la definición de dispositivo, en lugar de como dispositivos de diagnóstico autorizados. La frontera regulatoria para el AI clínico generativo y autónomo aún se está trazando, y los mercados que la dibujen primero (Corea hoy; otros que le seguirán) determinarán cómo los copiará el resto del mundo. Para los desarrolladores, la lección práctica es observar dónde se establecen los límites y diseñar la estrategia regulatoria, no sólo el producto, para un objetivo en movimiento.
El manual de acceso a los mercados
Si el problema es la fragmentación, la respuesta es un sistema. A través de los patrones anteriores, surge un manual repetible para llevar un dispositivo médico AI al mundo y mantenerlo allí.
1. Clasifique antes de construir la evidencia. Un mismo producto puede ser Clase II o Clase III dependiendo del mercado y del reclamo. Mapear las reglas de clasificación de los mercados objetivo primero, porque dictan la evidencia clínica y técnica que necesitará. Secuenciar la afirmación y las pruebas hasta el mercado objetivo más estricto evita reconstruir el expediente más adelante.
2. Obtenga una fuerte aprobación del ancla y luego aproveche la confianza deliberadamente. Una autorización FDA o una marca CE valen mucho más que un mercado; es la credencial que desbloquea rutas abreviadas en Singapur, Brasil, México, Australia, el Golfo y más allá. El arte es saber cuál ancla reconoce cada mercado objetivo y encaminar el expediente en consecuencia. Arabia Saudita, que exige validación local incluso de los AI aprobados en el extranjero, es un recordatorio de que la confianza es un mapa, no una manta.
3. Mantener la representación en el país donde sea obligatoria, que es la mayoría de los lugares. Un fabricante extranjero no puede tener su propio registro en Brasil (BRH), EU (Representante autorizado), China (agente legal), Japón (MAH/DMAH), Arabia Saudita, Emiratos Árabes Unidos, India y muchos más. Cada uno requiere una entidad legal local para mantener el registro y presentarse ante la autoridad sanitaria. Defender treinta entidades de este tipo no es práctico; subcontratarlos a un solo socio es la forma en que la escala se vuelve factible.
4. Trate el control de cambios como un flujo de trabajo multimercado de primera clase. Esta es la disciplina específica de AI. Construir un PCCP de EE. UU., pero también mapear cómo cada mercado maneja los lo mismo actualice y diseñe la cadencia de lanzamiento del algoritmo en torno al régimen más restrictivo que sea importante para su negocio. El plan de ciclo de vida ahora forma parte del plan de acceso al mercado.
5. Ejecútelo como una operación conectada, no como treinta presentaciones desconectadas. El costo no es un registro único; es la coordinación (traducciones, titulares locales, calendarios de renovación, notificaciones de cambios y vigilancia posterior a la comercialización) entre docenas de regímenes lo que va a la deriva de forma independiente.
Una secuencia típica en la práctica. Para un desarrollador de imágenes-AI, la implementación suele ser la siguiente: asegurar el FDA 510(k) o Marca CE como ancla; en paralelo, presente sus solicitudes en su mercado local y en un mercado de rápida dependencia (Singapur o Australia) para acumular ingresos anticipados; utilizar esas aprobaciones para abrir las rutas abreviadas en Brasil, México y el Golfo; luego, aborde los mercados de alto valor y alto esfuerzo — China y Japón — donde las pruebas locales o los datos clínicos son inevitables y los plazos de entrega son más largos. En todo momento, se mantiene centralmente un único plan de control de cambios y se asigna a las reglas de actualización de cada mercado, por lo que una mejora del modelo se presenta donde debe estar y en ningún lugar donde no necesita estar. El orden no es arbitrario; secuencia deliberadamente el flujo de caja, la reutilización de evidencia y los mercados de recursos escasos.
Este es el trabajo que Pure Global está diseñado para hacer: representación en el país y ejecución regulatoria asistida por AI en más de 30 mercados, entregado con una tarifa anual fija en lugar del modelo horario indefinido que utiliza la industria por defecto. Los datos de este informe, extraídos de FDA, EUDAMED, NMPA, PMDA, MFDS, ANVISA y docenas de registros nacionales, junto con nuestro propio conjunto de datos de cronograma y costos mercado por mercado, son la misma inteligencia que utilizamos para secuenciar el lanzamiento global de un cliente. El objetivo de mapear el laberinto de manera tan exhaustiva es poder guiar al cliente a través de él rápidamente.
Conclusión: cuatro cosas para llevar
AI es ahora una categoría de dispositivo convencional y los datos lo demuestran. De aproximadamente 1 de cada 700 autorizaciones de FDA en 2019 a 1 de cada 28 en 2025, con más de 1500 dispositivos AI autorizados solo en los EE. UU. y programas nacionales en Corea, China, Taiwán y Japón escalando rápidamente, AI SaMD ha pasado de la novedad a la norma.
Cada regulador lo clasifica mejor y la mayoría está convergiendo en la supervisión del ciclo de vida. Regla 11 en EU y Brasil, Clase III en China, alto riesgo según la Ley EU AI: el AI diagnóstico y terapéutico se trata como algo serio, y la respuesta compartida es el control del ciclo de vida total del producto, GMLP y planes de cambio predeterminados.
Reliance realiza un viaje de aprobación, excepto la parte AI. El mecanismo de armonización (IMDRF, MDSAP, rutas de dependencia) realmente permite que una aprobación de anclaje sólida desbloquee docenas de mercados. Pero el control de cambios adaptativo AI diverge marcadamente, por lo que una única aprobación no sigue siendo válida en todas partes a medida que evoluciona el modelo. Esa brecha es el desafío operativo definitorio del campo.
La ventaja competitiva es la máquina de registro, no el registro. Cuando las reglas difieren en cada mercado y cambian cada año, la ventaja duradera pertenece a aquellos que pueden registrarse rápidamente, en todas partes, y mantener viva cada aprobación a través de cada actualización de modelo, como un sistema conectado.
Habla con nosotros
Si está construyendo o ampliando un dispositivo médico AI y sopesando en qué mercados ingresar, en qué orden y cómo mantener válida cada aprobación a medida que su modelo mejora, ese es exactamente el problema que resolvemos. Habla con Pure Global sobre un plan de acceso al mercado basado en los datos anteriores, o explore nuestras guías de registro mercado por mercado para profundizar en un solo país.
Fuentes
Organismos gubernamentales y de armonización citados arriba; referencias clave agrupadas a continuación. Todas las cifras están fechadas; El tamaño del mercado y las cifras previstas son estimaciones de terceros cuyo alcance varía y deben leerse como rangos.
Definiciones, marcos y principios rectores
- IMDRF — SaMD: Definiciones clave (N10, 2013); Categorización de riesgos (N12, 2014); Dispositivos médicos habilitados para ML: términos clave (N67, 2022); Principios rectores del GMLP (N88, 2025); Manual de estrategias de confianza (N89, 2026). imdrf.org
- US FDA — Software como dispositivo médico (SaMD); Marco regulatorio propuesto para modificaciones a AI/SaMD basado en ML (2019); Plan de acción AI/ML (2021). fda.gov
- QUIÉN— Ética y gobernanza de AI para la salud (2021); Consideraciones regulatorias sobre AI para la salud (2023); Guía LMM (2024); Buenas prácticas de confianza, TRS 1033 Anexo 10 (2021). quien.int
Estados Unidos
- FDA — Dispositivos médicos habilitados por inteligencia artificial lista (actualización del primer trimestre de 2026, 1524 dispositivos; ~76 % radiología); Orientación final del PCCP (diciembre de 2024); Comité Asesor de Salud Digital en AI generativo (noviembre de 2024); Tarifas de MDUFA para el año fiscal 2026. fda.gov · The Imaging Wire (análisis de participación en radiología, 2026); Innolitics & IntuitionLabs (rastreadores de autorización, 2025-26).
Unión Europea y UK
- EUR-Lex — Reglamento (EU) 2017/745 (MDR), anexo VIII, regla 11; Reglamento (EU) 2024/1689 (Ley AI), artículos 6, 99, 113. Comisión Europea — MDCG 2019-11 Rev.1 y MDCG 2025-6. MedTech Europa y Equipo-NB (Capacidad del Organismo Notificado). MHRA: programa de cambio de software y AI; AI Esclusa de aire; CE: guía de reconocimiento. gov.uk
Canadá, Australia, Japón, Corea
- Salud Canadá — Guía previa a la comercialización de dispositivos médicos habilitados para ML (abril de 2026). canada.ca
- TGA — reformas de software (2021); Resultados de la consulta AI (2024-26). tga.gov.au
- PMDA/MHLW — Guía SaMD; GUIÓN para SaMD; IDATEN. pmda.go.jp
- MFDS — Informe de aprobación 2025 (153 dispositivos AI); AMPD; pauta generativa-AI. mfds.go.kr; bioin.or.kr
Asia-Pacífico, Latinoamérica, MEA
- HSA Singapur — GL-04-R4; agencias de confianza/referencia. hsa.gov.sg; moh.gov.sg
- NMPA China: clasificación AI y reforma de octubre de 2025. nmpa.gov.cn · JMIR Informática Médica (154 AIMD, 2026).
- CDSCO India: borrador de la guía de software MD (octubre de 2025). TFDA Taiwán: orientación CADe/CADx; J. Formos. Medicina. Asociación. (166 licencias).
- ANVISA Brasil — RDC 751/2022 (Regla 11), RDC 657/2022, IN 290/2024. gov.br/anvisa
- COFEPRIS México — Ruta Abreviada (2025). SFDA Arabia Saudita — MDS-G010 (2022). EAU EDE — Decreto-Ley Federal 38/2024. SAHPRA Sudáfrica — Comunicación AI (2025).
Mercado, empresas, patentes, seguridad.
- Tamaño del mercado: Grand View Research; Mercados y Mercados; La Empresa de Investigación Empresarial; Inteligencia de Mordor; Perspectivas empresariales de Fortune (2025-26).
- Financiamiento y empresas: Rock Health (financiación de salud digital 2025); Perspectivas de CB; presentaciones de empresas para el año fiscal 2025 (Tempus AI, iRhythm, HeartFlow, Butterfly Network, Lunit, VUNO); Aidoc (Serie E).
- Patentes: Tendencias tecnológicas de la OMPI (2019) y panorama generativo de patentes AI (2024); CNIPA.
- Seguridad: NEJM (sesgo del pulsioxímetro, 2020); JAMA Medicina Interna (Modelo de Sepsis Épica, 2021); JAMA Health Forum y JAMA Network Open (retirada de AI, 2025); Guía del pulsioxímetro FDA.
- Comercio: OCDE-FMI-OMC Medición del comercio digital (2021); OMC; UNCTAD.
- Conjunto de datos de cronograma y costo de acceso al mercado exclusivo de Pure Global (2026); Análisis de bases de datos openFDA, EUDAMED y MFDS: Pure Global, junio de 2026.



Hablemos,
esté donde esté.
Ya sea que busque más información o esté listo para asociarse con nosotros, le guiaremos en cada paso del proceso regulatorio.
Contáctenos