
医療機器としての AI: 規制、登録、市場アクセスの世界地図
人工知能は、歴史上最も急速に成長している医療機器であり、規制が最も多様です。米国の FDA には現在 1,524 台の AI 対応デバイスがリストされています。韓国は1年間に153件を認可した。しかし、同じソフトウェアが米国ではクラス II、EU ではクラス IIa+ および「高リスク」、中国ではクラス III であり、それぞれに独自の証拠、現地所有者、および変更管理ルールがあります。このレポートでは、各主要規制当局が AI を医療機器としてどのように分類、承認、規制しているかを、30 以上の市場にわたる登録コスト、タイムライン、信頼ルート、および毎回書類を再構築することなくそれらの規制に到達するためのハンドブックとともにマッピングしています。
世界の規制当局が医療分野における人工知能をどのように分類、承認、取り締まっているのか、また毎回書類を再構築することなく 30 以上の市場に参入する方法について説明した、証拠に基づいたフィールド ガイドです。
TL;DR
人工知能は、歴史上最も急速に成長している医療機器のカテゴリーとなっています。米国食品医薬品局の AI 対応デバイスの公開リストは 2026 年半ばまでに 1,524 件の認可に到達し、そのおよそ 4 分の 3 が放射線医学分野です (FDA)。 FDA 510(k) データベースを独自に分析したところ、全許可に占める AI の割合は 2019 年の約 700 件に 1 件から 2025 年には約 28 件に 1 件へ上昇しました。韓国だけでも 2025 年に 153 件の AI デバイスを認可しています (MFDS).
しかし、あるエージェンシーを通過する同じソフトウェアが、次のエージェンシーでも失速する可能性があります。このレポートの主張は次の 5 つの点にあります。
- AI はデバイスであり、そのほとんどは「医療機器としてのソフトウェア」(SaMD) です。 ソフトウェアが診断、トリアージ、または治療の推奨を行う場合、ソフトウェアはメスやスキャナーのように規制され、数十の管轄区域でそれぞれ独自の規則が定められています。
- 世界中がそれを上位に分類しています。 EU の MDR ルール 11、ブラジルのルール 11、中国の診断 AI のクラス III のデフォルト — 診断および治療用ソフトウェアは、ほぼどこでも、第三者によるレビューを必要とする高リスク クラスに分類されます。
- アダプティブ AI は 1 回限りの承認モデルを打ち破りました。 モデルは出荷後も変化を学習し続けるため、規制当局は米国の所定変更管理計画、日本のIDATEN、韓国のDMPAなどの新しいメカニズムを発明しました — しかし、これらは互いに一致しません。
- 信頼性は収束し、AI は発散します。 信頼性と認知度の経路が網の目状に形成されているため、1 回の承認で多くの市場が開拓できるはずです。静的デバイスの場合は機能します。しかし適応型 AI については、「FDA によって認可された PCCP は EU AI 法の義務を満たしておらず、その逆も同様です」(Berkley Lifesciences)。
- したがって、勝者は複数市場登録を産業化します。 競争力はもはや 1 つのクリアランスではありません。これは、1 つの承認を 30 に変換し、アルゴリズムが進化するにつれてそれぞれの承認を有効に保つ運用マシンです。
これは Pure Global のギャップを埋めるものです。国内代理店と AI が 30 以上の市場にわたる規制執行を定額の年会費で支援します。このレポートの残りの部分は地図です。
「医療機器としてのAI」とは一体何なのか
最も重要な役割を担う言葉、デバイスから始めましょう。ソフトウェアが「病気の診断、治療、予防に使用することを目的としている」場合、それは、MRI スキャナー内のチップ上で実行されるか、放射線科医のブラウザーのアプリとして実行されるかに関係なく、地球上のほぼすべての法制度において医療機器となります。 AI はほとんどの国で独自の法令を制定していません。それは医療機器法の仕組み全体を継承しています。
アンカーとなる定義は、主要な規制当局が用語をすり合わせる場である国際医療機器規制当局フォーラム (IMDRF) に由来します。 2013 年の基本文書で、IMDRF は医療機器としてのソフトウェア (SaMD) を「ハードウェア医療機器の一部ではなく、1 つ以上の医療目的で使用されることを目的とし、これらの目的を実行するソフトウェア」と定義しました (IMDRF N10)。 FDA はその言語をそのまま採用し、下流のすべてのものにとって重要な 3 方向の区別を示しています (FDA):
- SaMD — ソフトウェア です 医療機器 (胸部 X 線トリアージ アルゴリズム、糖尿病網膜症検出器)。ここは、臨床的な AI のほとんどが存在する場所です。
- SiMD — 医療機器のソフトウェア、ハードウェア (輸液ポンプを実行するファームウェア) に統合されています。
- 使用するソフトウェア 製造または保守 デバイスはまた違った方法で規制されます。
AI については特に、IMDRF の 2022 年の重要用語文書が機械学習対応医療機器を「意図された医療目的を達成するために、部分的または全体的に機械学習を使用する医療機器」と定義しています (IMDRF N67).
すべてを壊す区別: ロックとアダプティブ
従来のデバイス規制は、固定された設計についてデバイスが安全で効果的であることを一度証明すれば、その設計はそのまま維持される、という単純な取引に基づいています。 AI はその思い込みを打ち破ります。 FDA の 2019 年に重要なディスカッション ペーパーでは、この線が正確に描かれています。「ロックされた」アルゴリズムとは、「同じ入力が適用されるたびに同じ結果が得られ、使用しても変化しない」アルゴリズムです — ルックアップ テーブル、デシジョン ツリー、凍結された分類器などです (FDA、2019)。一方、適応型すなわち継続学習型のアルゴリズムは、導入後も変化し続けます。
この 1 つの特性、つまり現場で自らを改善するソフトウェアこそが、AI が 10 年間の新たな規制を必要とした理由です。 1 月に承認した製品が 6 月に病院で使用されていた製品ではない場合、一体何を承認したのでしょうか?このレポートのすべてのフレームワークは、結局のところ、その質問に答えようとする試みであり、 トータル製品ライフサイクル (TPLC) — クリアランスの瞬間だけでなく、デバイスの寿命全体にわたる監視 — が共通の対応です。
規制当局は見た目の厳しさをどのように決定するか
IMDRF の 2014 年のリスク分類フレームワークは、現在世界が従う論理を設定しました。SaMD の精査は 2 つの要素で評価されるべきです。 それが提供する情報の重要性(情報提供か、駆動か、それとも診断/治療か?)と医療状況の深刻さ(非深刻、深刻、または重大) (IMDRF N12)。腰痛のためのストレッチを提案するアプリと、脳出血にフラグを立てるアルゴリズムは同じ規制対象ではありません。この 2 軸グリッドがその理由です。
の 世界保健機関 倫理的な足場を追加しました。 2021年のレポート 健康のための人工知能の倫理とガバナンス 6つの原則を設定します—自律性を保護します。幸福と安全を促進する。透明性と説明可能性を確保する。責任と説明責任を促進します。包括性と公平性を確保する。応答性と持続可能性を促進します AI (誰が)。 WHO は、2023 年に規制に関する検討を続け、2024 年 1 月には、生成的な AI と大規模なマルチモーダル モデルを真正面から対象とした最初の世界的なガイダンスを発表しました (誰が).
私たちがここにたどり着いた経緯
AI 医療機器は、単一の画期的な進歩を伴って登場したわけではありません。それらはおよそ 10 年にわたって国ごとに蓄積されていきました。以下のマイルストーンは、2013 年の定義がどのようにして 5 大陸専用のライフサイクル ルールになったかを示しています。
AI 医療機器規制の世界的なタイムライン (2013 ~ 2026 年)
わずか 10 年の間に、AI SaMD は IMDRF の定義から 5 大陸専用のライフサイクル ルールに移行しました。
| 日付 | マイルストーン |
|---|---|
| 2013 年 12 月 | IMDRF N10 は「医療機器としてのソフトウェア」を定義しています (SaMD) |
| 2017 年 1 月 | FDA が Arterys をクリア — 初のクラウド + ディープラーニング臨床ツール (510(k)) |
| 2018年4月 | FDA が IDx-DR を承認 — 最初の自律的な AI 診断 (De Novo) |
| 2018年5月 | 韓国MFDSがVUNO Med-BoneAgeを承認 — 韓国初のAIデバイス |
| 2018年12月 | 日本 PMDA が EndoBRAIN (クラス III) を承認 — 日本初 AI SaMD |
| 2019年4月 | FDA AI/ML SaMD への変更に関するディスカッション ペーパー (ロックとアダプティブ) |
| 2020 | 中国 NMPA が DeepVessel FFR を承認 — 最初のクラス III AI デバイス。日本、SaMD 向けに IDATEN + DASH を開始 |
| 2021年1月 | FDA AI/ML アクション プラン。 2021年6月 WHOの6つの倫理原則 |
| 2021年10月 | GMLP — 10 の基本原則 (FDA + カナダ保健省 + UK MHRA) |
| 2022 | サウジ SFDA MDS-G010 — 初期の専用 AI デバイス ガイダンス (最初の「施行可能な」ものとして引用する人もいます)。 IMDRF N67 ML 用語。ブラジル RDC 751/657 |
| 2024 年 8 月 | EU AI 法律は発効します。 WHO LMM (generative-AI) ガイダンス (2024 年 1 月) |
| 2024 年 12 月 | FDA は、所定の変更管理計画 (PCCP) ガイダンスを最終決定します |
| 2025 年 1 月 | IMDRF N88 GMLP 決勝。韓国が世界初のジェネレーティブ-AI デバイスガイドラインを発行。 FDA AI ライフサイクル ドラフト ガイダンス |
| 2026 年 2 月 | IMDRF N89 信頼性ハンドブック |
| 2026 年 4 月 | カナダ保健省が ML デバイスのガイダンスを最終決定。韓国が最初の生成-AI デバイスを承認 |
出典: IMDRF、US FDA、EU、NMPA、PMDA、MFDS、SFDA、ANVISA、カナダ保健省およびWHOの一次文書 — Pure Global、2026年6月より編集。
いくつかの瞬間は強調する価値があります。で 2017年1月、FDA は Arterys をクリアしました — クラウド コンピューティングと深層学習を組み合わせた最初の臨床ツール (PRニュースワイヤー)。次に、 2018年4月、分水嶺がやって来ました: FDA が承認されました IDx-DR、最初の AI が診断の提供を許可されました 自律的に医師が結果を解釈することはなく、プライマリケアのための糖尿病網膜症スクリーニングは、その極めて重要な試験で感度 87.2%、特異度 90.7% に達しました (npjデジタルメディシン)。数か月以内に、韓国 (VUNO Med-BoneAge、2018 年 5 月) と日本 (EndoBRAIN、2018 年 12 月) が独自の初の薬を承認しました。中国は 2020 年に初のクラス III AI デバイスである DeepVessel FFR を発表しました (npjデジタルメディシン).
2021 年から 2025 年の間に規制の足場が整いました: 三極 優れた機械学習の実践 2021 年 10 月の原則、EU の AI 行為 2024 年 8 月に発効し、FDA の最終的な 事前に決定された変更管理計画 2024 年 12 月のガイダンス、そしてフロンティアの動きがどれほど速いかを物語る、韓国の 世界初の生成-AI デバイスガイドライン 2025 年 1 月 (バイオワールド)。 2026 年 2 月までに、IMDRF はグローバルな 信頼性ハンドブック 規制当局がお互いの仕事に頼れるよう支援するため(IMDRF N89).
データ: 大きさ、速度、場所
クリアランス曲線は急激に上向きに曲がっています
AI の医学への浸透を測る唯一の最良の尺度は、FDA の公開情報です。 人工知能対応医療機器 到達したリスト 1,524件の認可 最新の更新内容 (2026 年の第 1 四半期までのデータを反映) (FDA)。見るには 速度では、基礎となる FDA 510(k) データベースを直接分析しました。特定された AI/ML の許可は、2010 年代後半には少数でしたが、現在では増加しています。 2025年には115人 — そして、より明確に言えば、AI は 2019 年の全 510(k) 認可の 0.14% から 2025 年の 3.57%、6年間でシェアはおよそ25倍に跳ね上がりました。
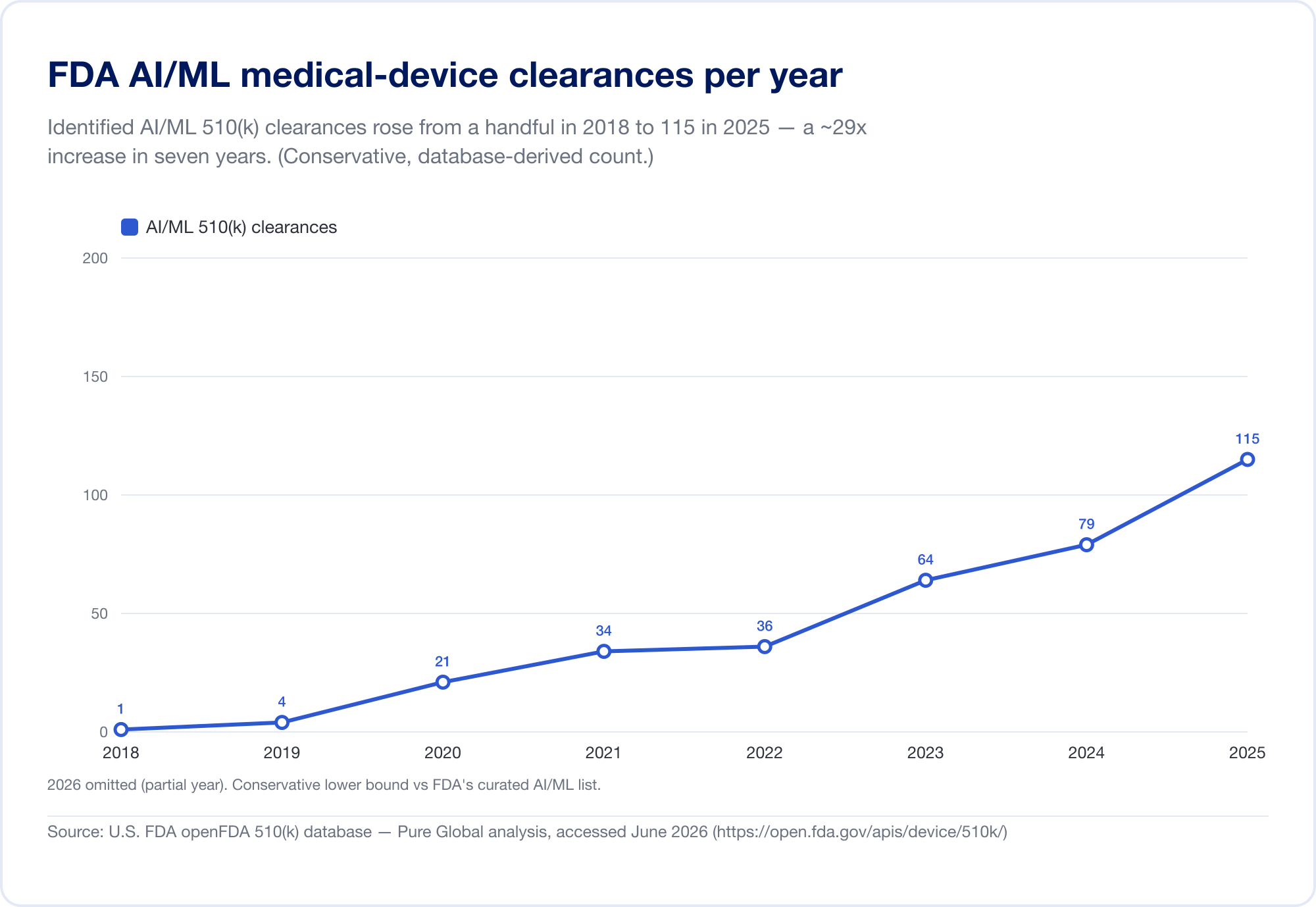
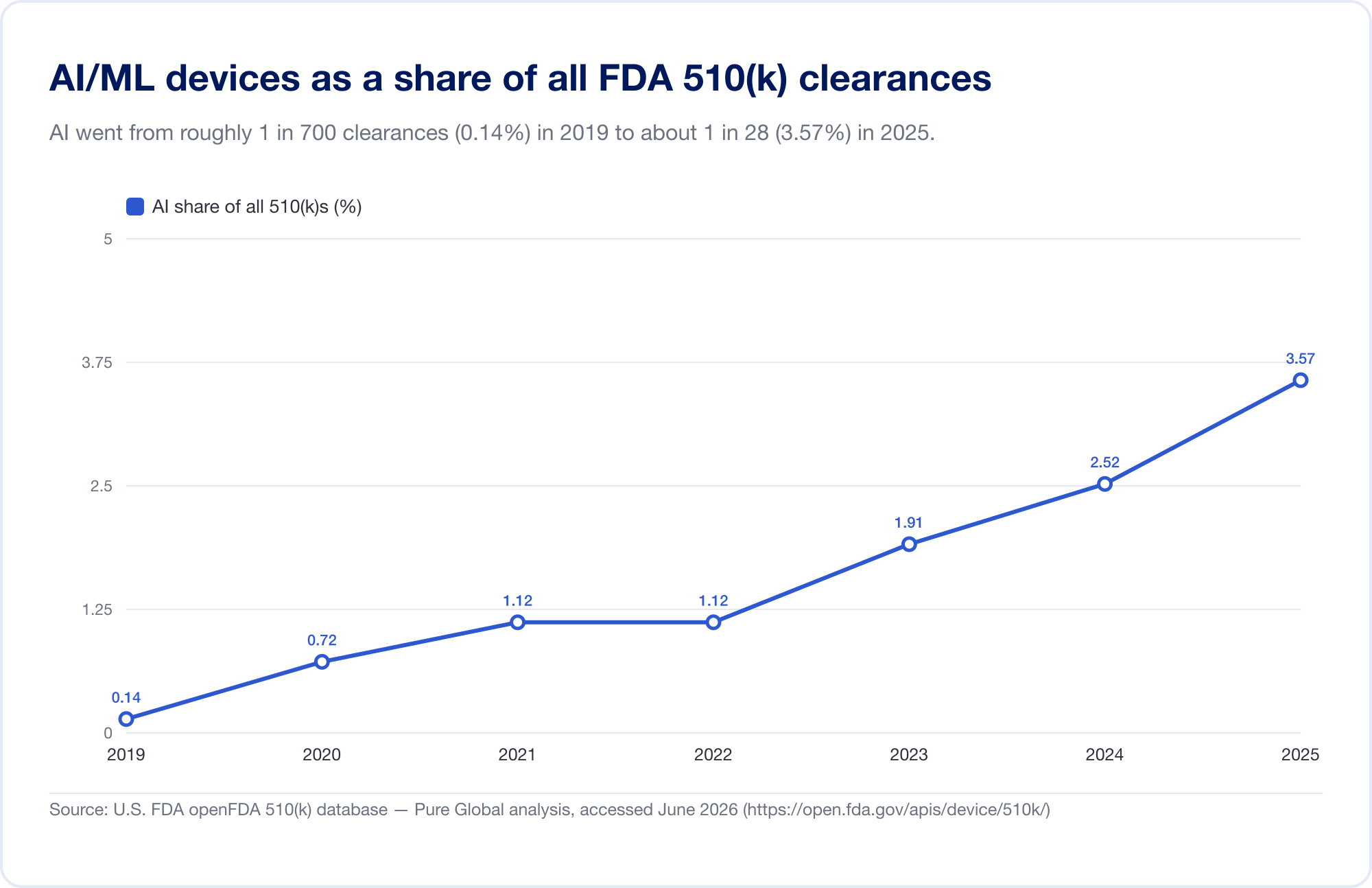
(データベースから導き出されたカウントは意図的に保守的であり、FDA の精選リストよりも狭いです。多くの AI 放射線科ツールはテキストに「AI」と決して書かれていない製品コードの下にあるためです。私たちはそれを傾向と地理に使用しています。FDA 独自のリストが見出しの合計です。出典: openFDA 510(k) データベース — Pure Global 分析、2026 年 6 月)
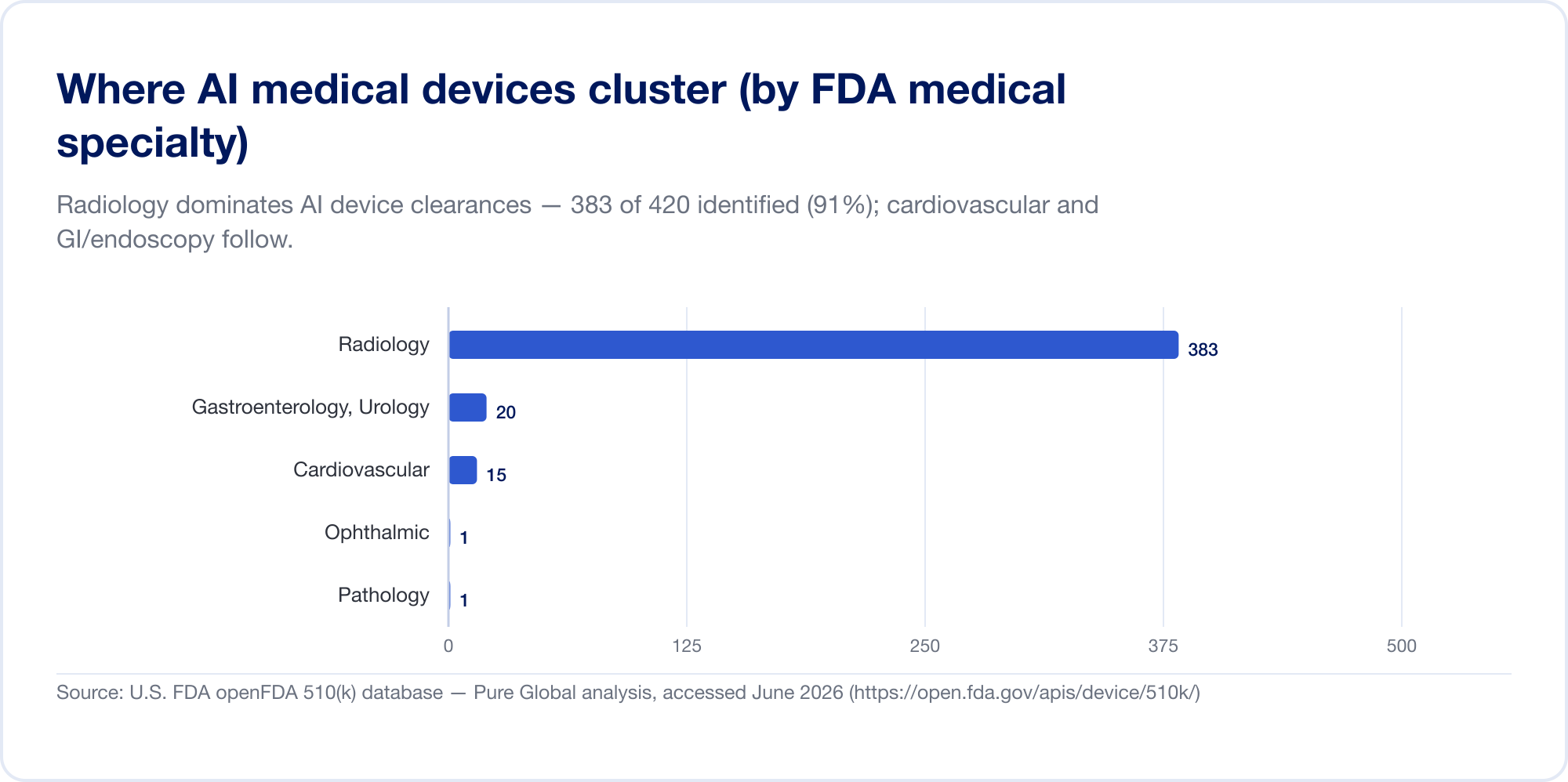
今のところ、それは圧倒的にイメージに関する話です
医学における AI は、今日では主に放射線医学における AI です。 FDA のリストでは、放射線科が占めています。 約76% すべての AI 承認のうち (イメージングワイヤー);私たち自身のクリアランスサンプルでは濃度はさらに高く、2位と3位は心臓血管病学と消化器病学(内視鏡検査)でした。その理由は構造的なものです。画像処理はデジタルで大量に行われ、ラベル付けされており、510(k) パスウェイにより、新しいアルゴリズムは既存のアルゴリズムを述語として引用できるようになります。病理学、心臓病学信号、および臨床テキスト モデルは成長していますが、重心は依然として画像です。
イノベーターはどこにでもいます。市場はどこにでもあります
ここに、商業化計画を再構築する必要があるという事実があります。 AI 米国で認可された医療機器は次のような国から来ています。 少なくとも16か国。 AI 510(k) の申請者の分析では、米国がトップですが、韓国、イスラエル、カナダ、フランス、中国、ドイツ、英国、オランダ、台湾、日本がそれぞれ意味のある数字を示しています。
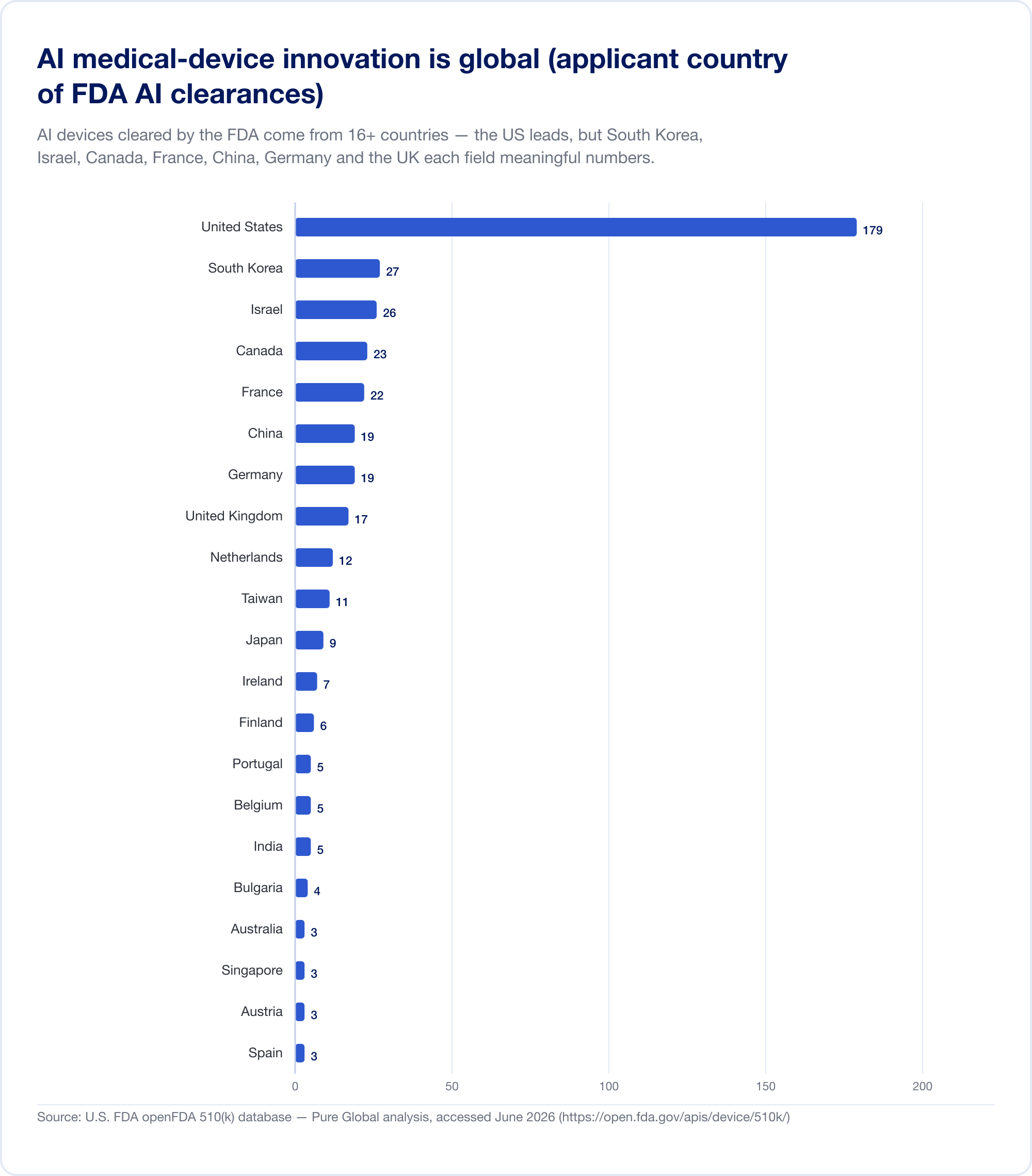
全国的な生産高はこのパターンを裏付けています。 韓国 認可済み (承認と認証を合わせた) AI デバイスは 2023 年に 64 台、2024 年には 108 台、 2025年には153人 — 41.6% 増加し、77.7% が国産 (MFDS). 中国 大まかに承認していました 154 AI 医療機器 2025 年半ばまでに、彼らの約 80% が最もリスクの高いクラス III (JMIR医療情報学). 台湾 2020 年から 2024 年までにライセンスを取得した 166 台の AI/ML デバイス (J. フォルモス。医学。准教授). 日本 2025 年 9 月の時点で、PMDA のリストには AI ベースの SaMD が 51 件ありました (世界の健康と医療).
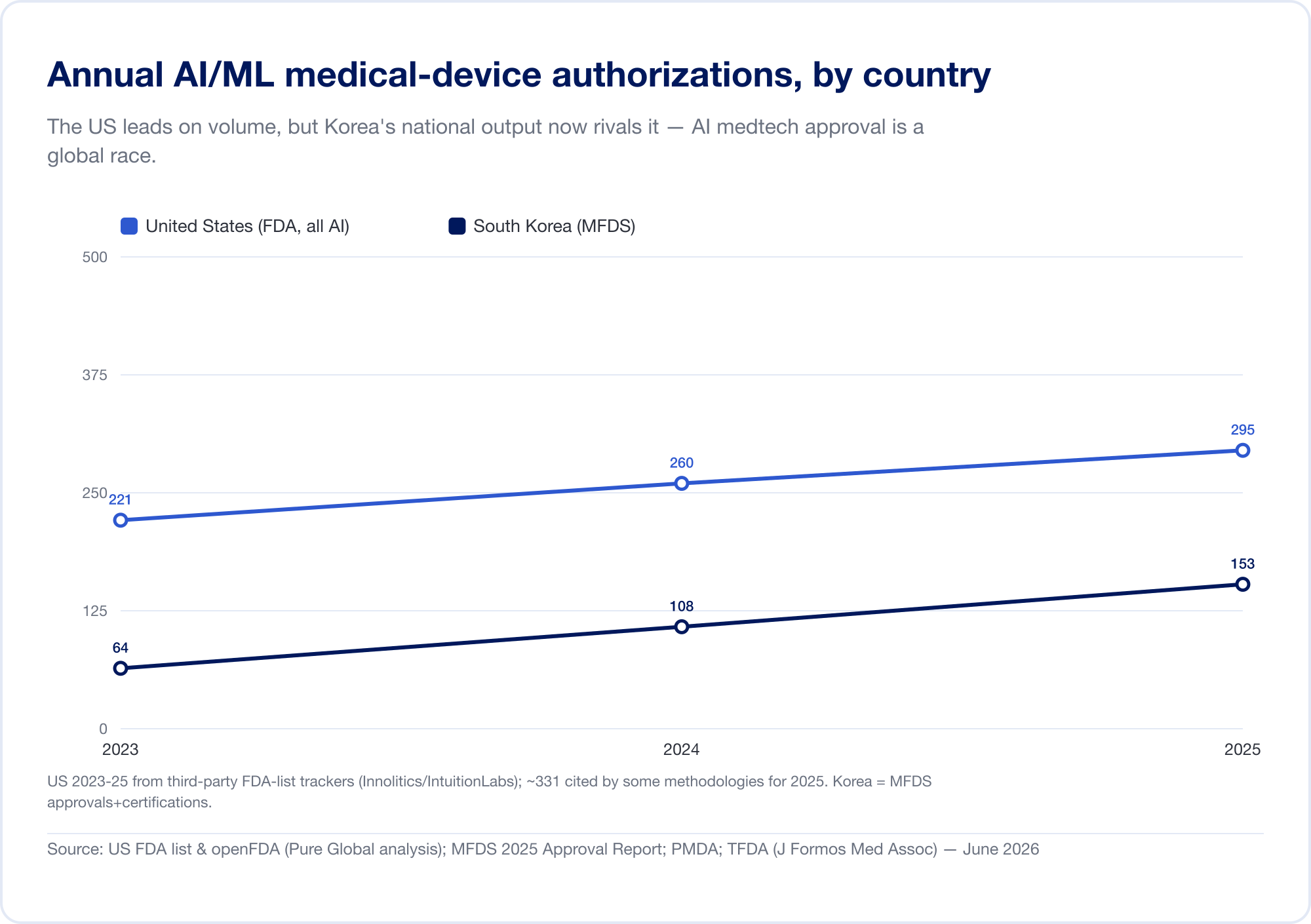
この意味は直接的です。テルアビブ、ソウル、上海で構築された優れたアルゴリズムは、それぞれが異なる規制言語を話す市場の患者に到達する必要があります。イノベーションはグローバルです。承認は頑固にローカルです。
市場の規模はどれくらいですか?それは完全にあなたが何をカウントするかによって異なります
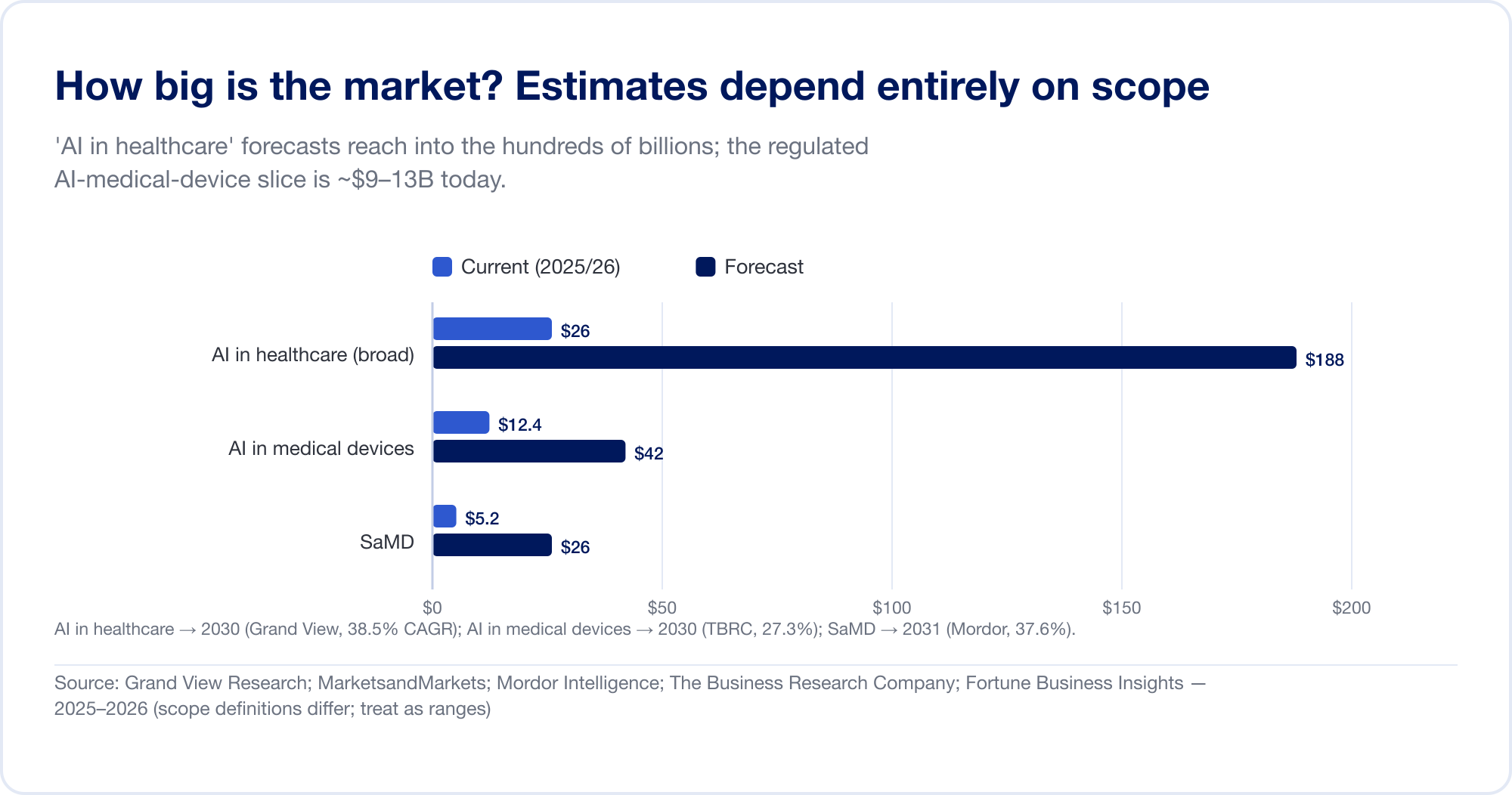
ヘルスケア分野における AI の市場規模の推定値は、アナリストが大きく異なる場所に境界線を引くため、桁違いに広がります。幅広い 「AI 医療分野」 創薬、管理自動化、アンビエントスクライブなどをバンドルしたカテゴリーは、次の時点で予測されています。 2030年までに1,877億ドル 38.5% の CAGR (グランドビューリサーチ)、最も積極的な住宅では 2034 年までに 1 兆米ドルを超えると予測されています (フォーチュン ビジネス インサイト)。より狭い、規制されたスライス — AI の医療機器 — はるかに小さく、より信頼性の高いものです: 2025年には124億ドル、2030年までに424億ドルに達する (ビジネスリサーチ会社)。純粋 SaMD 情報源にもよりますが、およそ 50 ~ 250 億米ドルと推定されています (モルドール・インテリジェンス)。これらすべてを事実ではなく範囲として扱います。
首都はもっときれいな物語を語ります。米国のデジタルヘルスベンチャーへの資金調達が回復 2025 年に 142 億ドル (+35%)、そして初めて AI 対応のスタートアップが過半数を獲得しました — 全ドルの54% (岩の健康)。 AI-SaMD の名前は、成長から収益性へと変化しています。 テンパス AI 2025 年度の収益は 13 億ドル (+83%) (テンパス); アイリズム 売上高は 7 億 4,700 万ドルに達し、GAAP ベースで最初の利益を計上した四半期 (アイリズム); ハートフロー 2025 年 8 月に上場し、40% 増加して 1 億 7,600 万ドルになりました (ハートフロー);韓国の ルニット 収益の 92% が海外からであり、53% 成長しました (ルニット)。個人プレイヤーの中には、 アイドック 2026年4月にシリーズEで1億5,000万米ドルを調達し、総資金調達額は5億ドルを超えた(アイドック).
特許競争には別のリーダーがいる
知的財産は、世界的な競争が最も顕著に表れる分野です。 WIPO の画期的な研究は大まかに数えられる 340,000 件の AI 関連の特許出願、生命科学と医学に次いで 3 番目に大きな応用分野 (ウィポ)。しかしその後地理が逆転し、2023 年までの 10 年間に、 中国は 38,210 件の生成的-AI 発明を出願しました - 世界の他の国々を合わせたよりも多いです — 対米国 6,276 (ウィポ)。中国特許庁は、2024 年末に専用の AI 特許審査ガイドラインを発行しました (クニパ)。医療 AI のパイプラインはアジアからの不均衡に満たされており、複数市場へのアクセス戦略が問題となっています いつではありません もし、世界の開発者の間でシェアが拡大しています。
盲点: 取引データに SaMD が表示されない
この市場についての考え方を再構築する分析上の注意点が 1 つあります。従来の医療技術市場のインテリジェンスは輸入と関税のデータに基づいていますが、 純粋な SaMD は見えません。クラウド ダウンロードまたはアプリ ストアによって配信されるソフトウェアは、物理的な国境を越えず、HS コードの税関申告も生成せず、商品貿易統計にも記録されません。国際的な標準設定者たちは、こう直接言います。 「現在の統計基準では、直接収益化されていないデータフローは一般的に取引フローとはみなされません。」 (OECD-IMF-WTO)、電子送信に対する WTO の関税一時停止措置は 1998 年以来行われています (アンクタッド)。 AI-組み込みハードウェア — AI 対応の CT スキャナー — 物品として移動し、データに表示されます。 510(k) をクリアしたクラウド アルゴリズムはそうではありません。結論: 従来の貿易分析を系統的に行う アンダーカウント AI ソフトウェアと過剰なハードウェア。 SaMD の世界的な広がりに関する唯一信頼できる足跡は、 登録 — これはまさにこのレポートが使用するレンズです。
安全計算
あらゆる規則強化の背後には、医療分野の AI が従来の機器では起こらなかった方法で失敗する可能性があるという一連の証拠があります。特に 3 つの調査結果は、規制当局の考え方を再構築しました。
プレーンなデータに潜むバイアス。 ランドマーク ニューイングランド医学ジャーナル 研究によると、パルスオキシメーター(遍在しており、アルゴリズムとの組み合わせが増えている)は、危険なほど低い血中酸素濃度(潜在性低酸素血症)を見逃していることが判明した。 黒人患者の 11.7% に対し、白人患者の 3.6%、約 50,000 のペアの読み取り値ではおよそ 3 倍の差異があります (NEJM、2020)。 FDA は 2021 年 2 月に安全性に関する通知を発行し、2025 年 1 月までに、肌の色調全体でのより多様な検証を要求するガイドライン草案を発行しました (FDA)。この教訓は一般化されました。非代表的な母集団でトレーニングされたモデルは、そのバイアスを導入するすべての病院に静かに影響を与える可能性があります。
現実との接触を生き延びない検証。 米国の数百の病院で導入されている独自の予測ツールであるエピック敗血症モデルは、38,455 件の入院を対象に外部検証され、最高のスコアを獲得しました。 曲線下面積は 0.63 で、ベンダーの主張する 0.76 ~ 0.83 をはるかに下回っています。。敗血症症例の 67% を見逃しましたが、全患者の 18% に対してアラートを発しました (JAMA内科、2021年)。モデルは大規模に「運用中」であっても、宣伝どおりに機能しない場合があります。
クリア直後のあのクラスターを思い出す。 2025 年のジョンズ ホプキンス大学とイェール大学の FDA 承認済み AI デバイスの分析では、次のことが判明しました。 AI デバイスのリコールの 43.4% は、認可されてから最初の 12 か月以内に発生しました。これは、510(k) デバイス全体の約 2 倍です。 (JAMA 健康フォーラム、2025 年)。並行して行われた調査では、公表された臨床研究のない機器にリコールが集中していることが判明した(JAMAネットワークオープン、2025年)。コンテキストが重要: おおよそ AI/ML デバイスの 97% が 510(k) 経路経由でクリア将来に向けた人体検査は必要ありません。そのため、市販後の警戒に大きく依存します。
安全計算: 規制当局が AI に対する規制を強化している理由
バイアス、検証のギャップ、早期リコールは、ライフサイクル監視への移行の背後にある証拠です。
| 見つける | 図 | ソース |
|---|---|---|
| パルスオキシメーターによる潜在的低酸素血症、黒人患者と白人患者 | 11.7% 対 3.6% (約 3 倍) | NEJM、2020 年 12 月 |
| エピック敗血症モデルの外部 AUC (vs 0.76 ~ 0.83 と主張) | 0.63;敗血症症例の67%を見逃した | JAMAインターン。医学、2021 年 6 月 |
| AI-認可から 12 か月以内にデバイスがリコールされる | 43.4% (すべての 510(k) の約 2 倍) | JAMA 健康フォーラム、2025 年 |
| AI デバイスがリコールされました (903 件が調査されました) | 4.8%、臨床研究が不足している患者に集中 | JAMAネットワークオープン、2025年 |
| AI デバイスは 510(k) 経由でクリアされました (将来的なテストは必要ありません) | ~97% | FDA リストの分析、2025 |
これにさらに次の問題が加わります データセットのシフト — 導入されたモデルは、患者集団、スキャナー、または周囲のコーディング システムが変化するにつれて静かに劣化します — そして、優れた機械学習の実践、所定の変更管理計画、および必須の現実世界のパフォーマンス監視という、最新の装置全体の理論的根拠が得られます。 AI が機能しないため、規制は強化されていません。効くから締めるんだよ 静かにそうならなくなるまで.
世界的な規制マップ
これはレポートの参照核心であり、2026 年半ばの時点で主要な管轄区域が AI SaMD を実際にどのように分類、審査、取り締まっているのかを示しています。視聴するスルーラインは 分類 (ソフトウェアがどのリスク クラスに分類されるか) および 変更制御 (アルゴリズムが更新されると何が起こるか)。統合された比較マトリックスは、地域の詳細に続きます。
15 の管轄区域が AI を医療機器としてどのように規制するか (2026 年)
同じソフトウェア、15 の回答: 分類、AI 固有のガイダンス、および変更管理ルールは市場ごとに異なります。
| 管轄区域 | AI SaMD が最も多く集まる場所 | 専用の AI/SaMD ガイダンス | 変更制御/適応型AIメカニズム | 外国の承認への依存 |
|---|---|---|---|---|
| 米国 (FDA) | クラス II (510(k)/デノボ) | はい — PCCP 最終 2024;ライフサイクル ドラフト 2025 | PCCP (変更の事前承認、新規送信なし) | いいえ (独自のレビュー、述語ベース) |
| 欧州連合 | クラス IIa+ (MDR ルール 11) + AI 高リスクの行為 | MDR + AI 法 + MDCG 2025-6 | 大幅な変更 → 認証機関の再審査 + AI 法 | いいえ (CE 適合性評価) |
| イギリス (MHRA) | クラス IIa+ (UK MDR 2002) | ソフトウェア & AI 変更プログラム; AI エアロック | PCCP 計画 (法定手段) | CE は 2028/2030 年まで GB で受け入れられます |
| カナダ (カナダ保健省) | クラス II ~ IV | はい — ML ガイダンス最終版 2026 年 4 月 | PCCP | MDSAP QMS の場合;完全に製品に依存しているわけではない |
| オーストラリア (TGA) | クラス IIa ~ III | AI レビュー 2024 (14 件の結果) | 開発中 | 同等の海外規制当局ルート |
| 日本 (PMDA/厚生労働省) | クラス II ~ III | SaMD の場合はダッシュ。 SaMD 案内 | IDATEN (PACMP) の事前合意された変更 | 外国の臨床データを受け入れます。 MDSAP |
| 韓国 (MFDS) | 2 ~ 3 年生 | はい — 含む世界初の生成-AI ガイドライン | DMPAの事前承認された変更計画 | 限定的;自分自身のレビュー |
| シンガポール (HSA) | クラスB~D | はい — GL-04-R4 (2025)、AI-MD ライフサイクル | 変更通知 | はい — 5 つの参照機関 (約 98% が要約) |
| 中国 (NMPA) | クラス III (判定ソフトウェア) | はい — CMDE AI 原則 + 分類カタログ | コアアルゴリズムが変更されていない場合のみカーブアウト | いいえ (国内代理店、型式試験) |
| インド (CDSCO) | クラスA~D | ドラフトのみ (2025 年 10 月) | アルゴリズム変更プロトコル(案) | 参照国の承認によりクラス C/D が緩和される |
| 台湾 (TFDA) | クラス II ~ III | はい — CADe/CADx + PCCP 製図ガイダンス | PCCP ガイダンス (2024) | ローカルパフォーマンス評価を重視 |
| ブラジル (ANVISA) | クラス II ~ IV (規則 11) | RDC 657 (SaMD);専用の AI ルールはありません | 完全変更登録 | はい — IN 290/2024 (クラス III/IV、4 機関) |
| メキシコ (COFEPRIS) | クラス I ~ III | デバイスの一般的なルール | 再登録 | はい — 省略されたパス (IMDRF + MDSAP) |
| サウジアラビア (SFDA) | クラスA~D | はい — MDS-G010 (初期; 最初の「強制可能」として引用) | GHAD経由の変更通知 | サポートのみ。ローカル検証が必要です |
| アラブ首長国連邦 (EDE) | クラス I ~ III | デバイスの一般的なルール | 再登録 | はい — CE/FDA を認識します |
出典: US FDA、EU MDR/AI 法、MHRA、カナダ保健省、TGA、PMDA/MHLW、MFDS、HSA、NMPA、CDSCO、 TFDA、ANVISA、COFEPRIS、SFDA、EDE — Pure Global 分析、2026 年 6 月。
米国 — ベンチマークであり、最も忙しい国
米国は世界で最も活発な AI デバイス体制を運営しており、FDA のデバイスおよび放射線医療センターとそのデジタル ヘルス センター オブ エクセレンスによって管理されています。特別な「AI 法令」はありません。デバイス定義を満たす AI 関数は、次の 3 つの経路を通じて SaMD として規制されます。 510(k) クリアランス (述語装置との「実質的同等性」を実証)、 デノボ 分類 (述語のない低リスクから中リスクの新規デバイスの場合)、および PMA (市販前承認、最もリスクの高いクラス III 用)。 AI デバイスの圧倒的多数 - 約 97% — 510(k) 経由で入力します。 De Novo を使用したことがあるのは数十人だけで、PMA は少数です (FDA リストの分析)。 De Novo のランドマークは 2018 年の IDx-DR でした。
最近の発展を特徴づけているのは、 事前に決定された変更管理計画 (PCCP)、2024 年 12 月に最終決定 (FDA)。 PCCP を使用すると、メーカーは将来の一連のモデル変更を事前に指定し、事前に承認できます。そして、メーカー自身の言葉がその価値を捉えています。FDA が PCCP をレビューします。 「各変更を実装するために追加のマーケティング申請を必要とせずに、デバイスの継続的な安全性と有効性を確保するため。」 2025 年 1 月、FDA はさらに前進し、AI 対応デバイス ソフトウェアの製品ライフ サイクル全体に関する包括的なガイダンス草案を発行しました。米国の姿勢を一言で言えば、高速、述語主導、イメージング重視、そして現在はライフサイクル監視を中心に組織化されています。
欧州連合 — 2 つの体制を 1 台のデバイスにスタック
EU は、2 つの規制制度が同時に適用されるため、AI SaMD にとって最も困難な主要市場です。まず、 医療機器規制 (MDR)。そのソフトウェア分類ルール — 規則 11 —悪名高いです。直接読んでください: 「診断または治療目的で意思決定を行うために使用される情報を提供することを目的としたソフトウェアはクラス IIa に分類されます。」 にエスカレートする クラスIII 誤った決定が死亡または不可逆的な悪化を引き起こす可能性がある場合、または クラス IIb 深刻な劣化の場合(MDR、付録 VIII、EUR-Lex 経由)。古い指令では、ほとんどのスタンドアロン ソフトウェアがクラス I として自己認定されており、第三者は関与していません。ルール 11 により、SaMD の診断および治療のほぼすべてが次の基準に達しました。 クラスIIa以上、これは強制的に 認証機関 適合性評価、ISO 13485 品質管理、および臨床評価。 EUDAMED データベースを独自にスキャンしたところ、AI キーワード デバイスがクラス IIa (まさにルール 11 のフットプリント) に集中していることがわかりました。
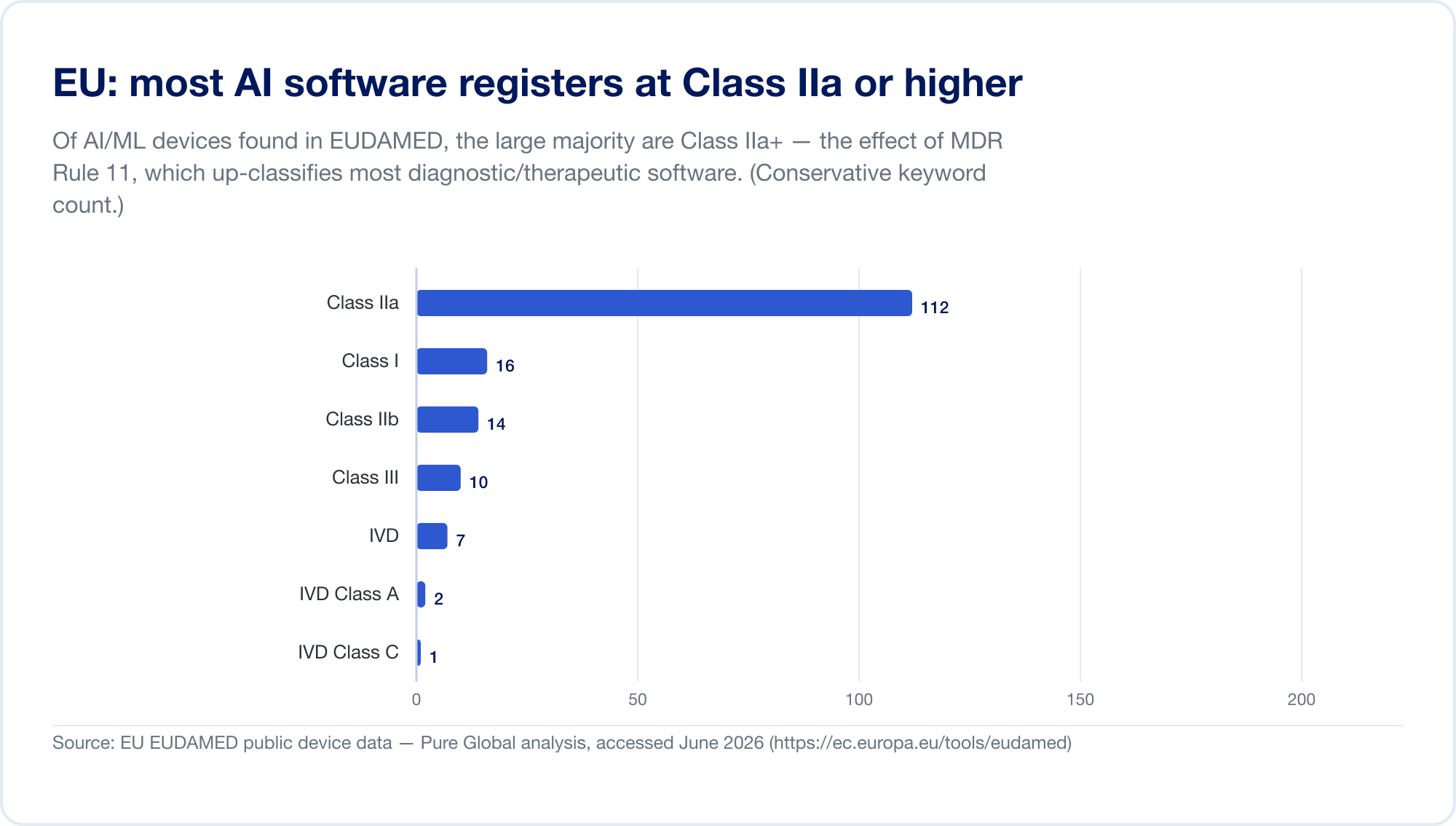
そして、その上に重ねて、 EU AI 法 (規則 2024/1689)、2024 年 8 月 1 日から施行されます。第 6 条(1)により、AI システムは 「ハイリスク」 それがすでにサードパーティによる適合性評価を必要とする製品 (またはその安全コンポーネントである) の場合、これには基本的にすべてのクラス IIa+ AI 医療機器が含まれます。高リスク義務は 2026 年 8 月 2 日まで段階的に導入され、MDR/IVDR デバイスに埋め込まれた AI の日付が 2026 年 8 月 2 日に設定されます。 2027 年 8 月 2 日 (2025 年 11 月の「デジタル オムニバス」提案では、これが 2028 年に延期される可能性があります。日付は移動したものとして扱われます) (人工知能act.eu)。罰則は次のとおりです 3,500万ユーロ、または世界売上高の7% (第99条)。重複を明確にするために、MDCG と新しいヨーロッパの AI 理事会は、2025 年 6 月に共同 FAQ MDCG 2025-6 を発行しました (欧州委員会).
バインド制約は容量です。 MDR 認証機関の数は、この指令により約 80 ~ 96 から約 80 に減少しました。 50、IVDR の下で指定されるのは ~17 ~ 19 だけです。 MDR 認証が取得されました 平均13~18か月、MDR 以前の標準のおよそ 2 倍 (MedTech ヨーロッパ)。 AI SaMD の場合、すべてのデバイスが希少な認証機関スロットをめぐって競合します。2027 年から 2028 年以降は、AI 法への適合性評価も必要になります。
英国 — 現実的に分岐している
Brexit 後、MHRA は意図的にイノベーションに優しい方針を打ち出しました。それ ソフトウェアと医療機器変更プログラムとしての AI (ロードマップは 2022 年 10 月に発行) は 11 の作業パッケージに及び、当局は今後の市販前規則で PCCP を許可することを約束しました (MHRA)。それ AI エアロック AI 医療機器向けのこの種のものとしては初めての規制サンドボックスは、2024 年に 4 つのプロジェクトのパイロットを実施し、2026 年まで 7 つのテクノロジーのフェーズ 2 を実施し、現在複数年にわたる資金提供が約束されています (MHRA)。実際、英国市場に出回っているデバイスの約 90% には依然として CE マークが付いており、英国はこのマークを受け入れることになります。 2028–2030; MHRA は 2026 年初めに CE マークを無期限に認識することについて協議しました (MHRA).
カナダ — 専用の ML ルールを最初に最終決定
カナダ保健省は、最初の規制当局の 1 つでした。 確定した 機械学習対応医療機器専用の市販前ガイダンス — 2025 年に初めて最終決定され、2025 年に改訂された最終形式で公開されます。 2026年4月 — クラス II ~ IV をカバーし、IMDRF の主要条件を採用し、承認された変更によって新たなライセンス修正が引き起こされないよう PCCP を正式に導入します (カナダ保健省)。カナダは、GMLP (2021)、PCCP 原則 (2023)、透明性原則 (2024) という 3 つの規制に関する基本文書を共同作成し、2019 年から MDSAP 認証を要求しています。
オーストラリア — 早期に改革し、AI に合わせて再調整中
オーストラリアの TGA は 2021 年 2 月にソフトウェア規則を改正し、低リスクのウェルネス アプリを切り出し、診断ソフトウェアを上位分類しました (診断機能を備えた治療用のアクティブ デバイスはクラス III に移動しました)。 2024年の協議では、 AIの規制の明確化と強化、600 人を超える関係者が参加し、14 の重要な発見が得られ、現在検討中です (TGA)。 TGA の依存ルート、つまり「同等の海外の規制当局」を受け入れるというルートは、以下で説明するように、大きな加速要因となっています。
日本 — 反復のために構築
日本は SaMD を「プログラムされた医療機器」として規制しており、おそらく主要市場の中で最も AI アップデートに優しいメカニズムを備えています。 いだてんは、2020 年 9 月から施行されており、承認後変更管理プロトコルの日本版であり、メーカーが頻繁に更新される AI (PMDA)。と組み合わせると、 SaMD のダッシュ イニシアチブと先駆け優先経路により、日本は進化するソフトウェアのインフラストラクチャを構築してきました。ただし、その導入はわずかであり、2025 年 9 月現在、PMDA のリストには AI ベースの SaMD が 51 件しかありません。
韓国 — 最も早い動き
韓国が顕著だ。 2025 年までに 153 件の AI の認可を取得したほか、目的に応じた法的枠組みを構築しました。 デジタル医療製品法 (DMPA)2025 年 1 月より発効し、PCCP スタイルの変更メカニズムと、IMDRF 作業項目に合わせたデジタル QMS が導入されます (エマーゴ)。韓国も発行した 世界初の発電用ガイドライン -AI 医療機器 2025 年 1 月に承認され、2026 年 4 月に最初のそのようなデバイスが承認されました。また、IMDRF AI/ML ワーキング グループの議長を務めています。世界的な AI 規制がどこへ向かうのか知りたければ、ソウルを見てください。
シンガポール — 信頼の拠点
シンガポールの HSA は、最も効率的であるため、その規模をはるかに超えています。 依存 アジアの政権。そのソフトウェアのガイダンス、 GL-04 (改訂 4、2025 年 12 月)、ライフサイクル全体にわたる機械学習対応デバイスを明示的にカバーしており、AI モデルのパフォーマンス、入力、または人間の監視レベルが変更された場合には変更通知が必要です (HSA)。重要なのは、HSA が 5 つの参照機関 (US FDA、EU 認証機関、カナダ保健省、TGA、日本の厚生労働省) を認めており、 約 98% のアプリケーションが短縮ルートを使用できると推定されています;事前に 2 つの承認を得たデバイスは、「即時」ルートを通じてわずか 1 時間で登録できます (シンガポール保健省).
中国 — 大きく、独特で、要求が厳しい
中国の NMPA は、AI 意思決定支援ソフトウェアを真剣に扱っています。2021 年の CMDE レビュー原則と、診断を提供したり治療を推進したりする 2021 ~ 2022 年の分類カタログを配置するソフトウェアです。 クラスIII、最も高いリスク層。中国は 2020 年に最初のクラス III AI デバイスを承認し、おおよその目標に達しました 2025 年半ばまでに 154 AI 医療機器 (JMIR医療情報学)。 2025 年 10 月の改革パッケージで、NMPA は次のことを約束しました。 「AI を搭載した医療機器の変更登録要件を簡素化します。コア アルゴリズムは変更されず、アルゴリズムのパフォーマンスは最適化されます。」 — 米国の PCCP と比較すると実質的だが狭い譲歩 (NMPA)。中国は国内代理店、現地での型式検査、そして多くのクラス III 機器については現地での臨床データを必要としており、このレポートの中で最も要求の厳しい市場の一つとなっています。
インド、台湾、その他のアジア太平洋地域
インドの CDSCO は 2025 年 10 月に医療機器ソフトウェア ガイダンスの草案を発行し、AI 更新のための「アルゴリズム変更プロトコル」を導入しましたが、まだ最終的な AI ルールが存在しない草案のままです (CDSCO). 台湾の 対照的に、TFDA は、どこよりも深い AI ガイダンス スイートの 1 つ (専用の CADe/CADx 技術ガイドラインと PCCP 製図ガイダンス) を備えており、2020 年から 2024 年までに 166 台の AI/ML デバイスがライセンス供与されています。 ASEAN 全体のパターンは、信頼性とローカリゼーションです。 マレーシア、タイ、ベトナム、フィリピン、インドネシア ほとんどの企業は SaMD を一般的なデバイスとして扱い、参照国の承認に依存していますが、ベトナムのファストトラックは異常に広範囲です (NMPA や MFDS の承認も受け入れています)。
ラテンアメリカ、中東、アフリカ
ブラジルの ANVISA が EU のロジックを大規模にインポートしました: 規則 11 RDC 751/2022 は EU を反映し、意思決定支援ソフトウェアをクラス II ~ IV に分類し、RDC 657/2022 はこの地域初の SaMD 固有の解決策でした。外国の製造業者はブラジルで登録を行うことはできません。 ブラジル登録者 は必須であり、放棄することはできません (アルティシオ). メキシコの COFEPRIS は 2025 年に依存体制を全面的に見直し、すべての IMDRF と MDSAP のメンバーを認める単一の短縮パスウェイとなり、30 営業日を目標としました。 サウジアラビアの SFDA発行 MDS-G010 (2022 年 11 月) — どこでも最初の AI/ML 専用の医療機器ガイダンスの 1 つであり、一部の観察者によって最初のものとして引用されています 強制力のある 1 つ (他の人は拘束力がないと分類しています) — それを独自に指示します。 「メーカーは、他の管轄区域で開発および承認された AI/ML ベースの医療機器を現地で検証する必要があります。」 (SFDA) — 「参照国の承認」だけでは必ずしも十分ではないことを思い出させます。の アラブ首長国連邦 2025年1月に新たなエミレーツ医薬品施設の下で一元的に機器の承認が行われます。 南アフリカの SAHPRA は 2025 年 9 月に最初の AI 通信を発行しましたが、まだデバイスの登録を開始していませんでした。 アフリカ医薬品庁 — 55 州中 31 州が批准 — まだデバイスまたは AI をカバーしていません。
これが問題の核心です。15 の管轄区域と 15 の回答です。同じソフトウェアは、米国ではクラス II、EU ではクラス IIa+ および「高リスク」、中国ではクラス III、韓国ではグレード 2 ~ 3、ブラジルではクラス II ~ IV であり、それぞれに独自の証拠、言語、現地所有者、および変更管理要件があります。
費用と所要時間
上記の分類の違いは、金額と月数に直接変換されます。以下の見出し番号は、 政府手数料と現実的なスケジュール リスクの高いデバイスの場合。これらには、公的な手数料に比べて見えにくい検査、臨床証拠、翻訳、国内代理にかかる多額の費用が含まれていません。
公式手数料は少額です
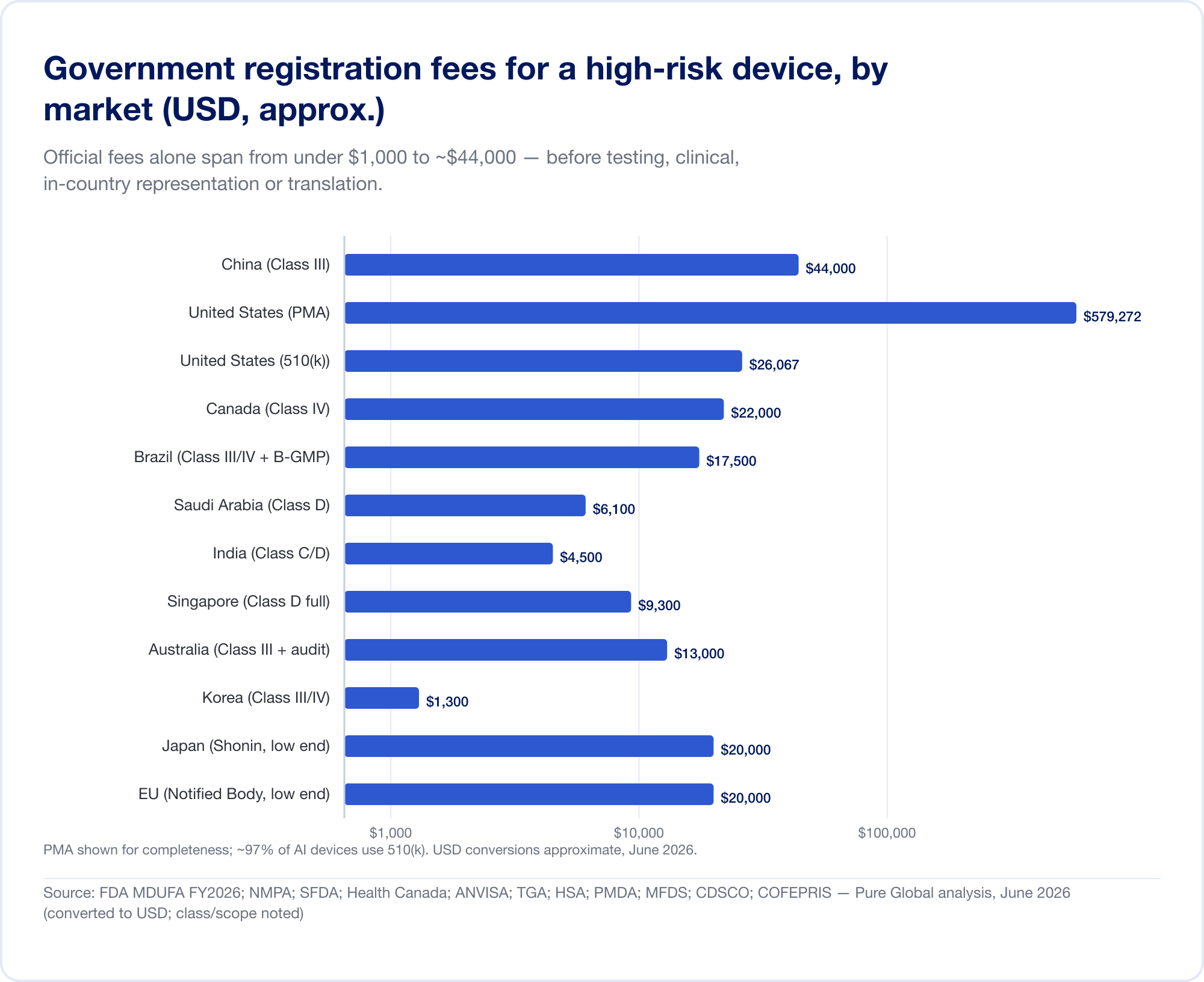
高リスク登録の政府手数料だけでも、1,000 米ドル未満から約 44,000 米ドルまでの範囲があります。
- 米国 — MDUFA 2026 年度手数料 (FDA.gov で確認): 510(k) $26,067 (中小企業 $6,517);デノボ $173,782; PMA $579,272;プラス年間設立費 $11,423 (FDA).
- 中国 — NMPA 登録料はおよそ 210,900人民元 (~30,000ドル) クラス II および 308,800人民元 (~44,000ドル) クラス III の場合 — このレポートで最も高額な公式手数料。
- ブラジル — ANVISA クラス III/IV の登録は ~21,000 レアル、さらに B-GMP の認定料がかかります レアル 72,804 海外メーカー向け。
- カナダ — クラス III CAD $14,163、クラス IV CAD $30,713 (2026 年 4 月)。
- サウジアラビア — SFDA手数料 15,000 ~ 23,000 サランドリ クラスごとに。
- インド — MD-15輸入ライセンス サイトあたり 3,000 ドル + 製品あたり 1,500 ドル クラスC/D用。
- シンガポール、オーストラリア、韓国、日本 — 公式手数料は比較的少額ですが(多くの場合 13,000 米ドル未満)、証拠と審査の負担は大きく異なります。
時間は高価な数字だ
実際にかかる費用はカレンダーです。現実的な高リスクのスケジュールは、約 1 か月 (カナダ、クラス III) から EU で 24 か月 そして 中国に4~5年 現地での臨床試験が必要な場合:
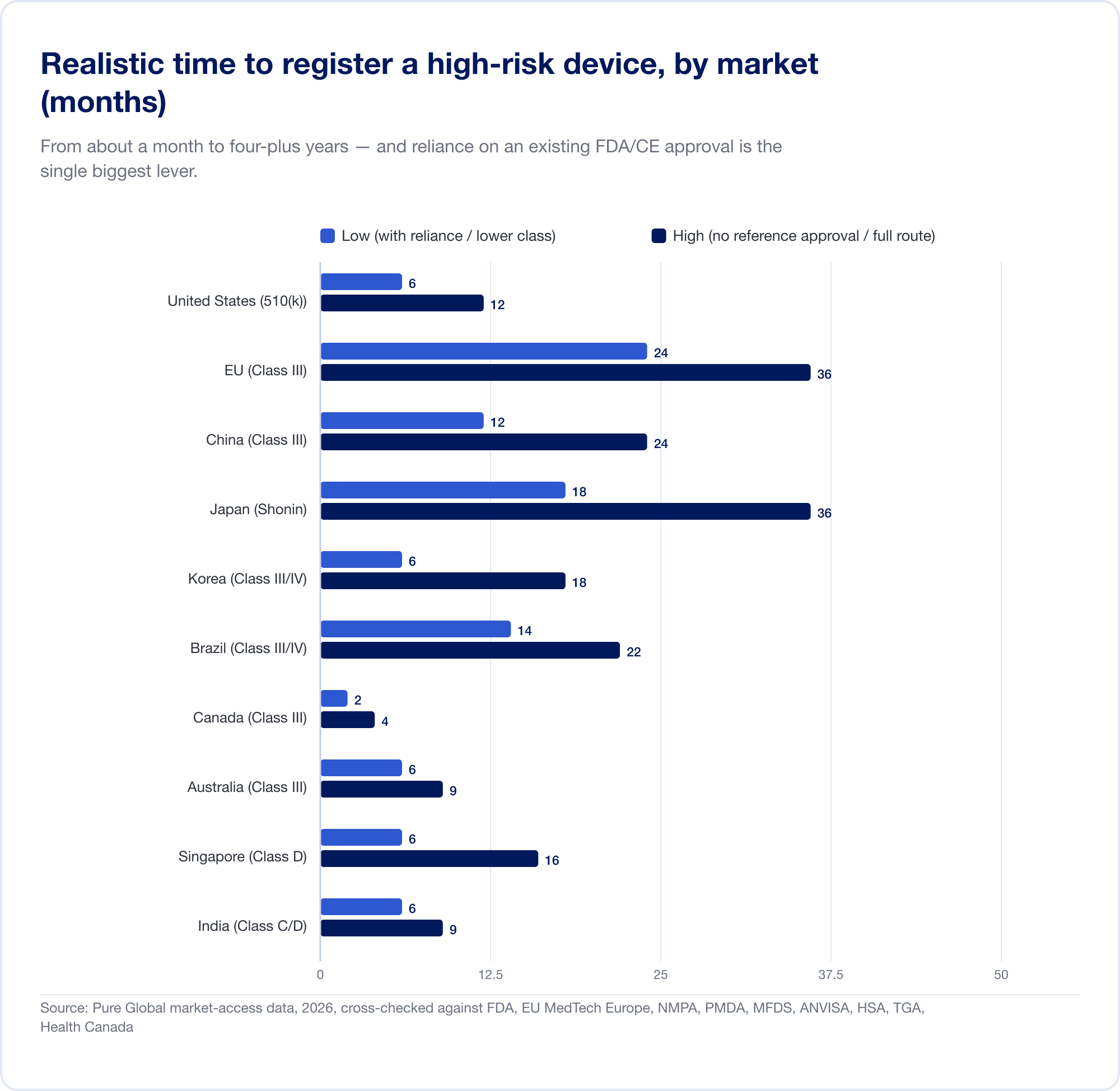
EU は、分類がどのようにコストになるかを示しています。ルール 11 は、これまでほぼ無料のクラス I 自己宣言であったものをクラス IIa+ 認証機関の関与に変換するため、SaMD のオールイン CE プロジェクトは通常 6 桁になり、 13 ~ 18 か月 証明書が発行される前に。四半期単位の滑走路を持つスタートアップにとって、それは項目ではなく、戦略的な脅威です。
レバー: 信頼と AI 特有のひねり
これらのタイムラインに対して、世界市場へのアクセスにおいて最も強力な唯一のツールが存在します。 依存。米国、EU、中国以外のほとんどの市場は、すでに取得している承認に依存します。当社独自の市場データは、その影響を顕著に示しています。標準ルートでブラジルで登録するのに約 8 か月かかる高リスクの SaMD は、 6週間 FDA またはその他の参照承認が ANVISA の最適化された分析経路を通じて利用される場合。
AI SaMD には、コスト表には示されていない特定の繰り返しのひねりがあります。 AI が常に更新しているように、アルゴリズムが更新されると、問題はその更新に更新が必要かどうかです。 新しい 提出。米国の PCCP では、事前に指定されたアップデートを新たに提出することなく出荷できるため、 26,067 ドルの料金と 90 ~ 175 日間の審査 回避された 510(k) ごとに、回避された PMA サプリメントではさらに多くの値が得られました。について FDA が 2025 年に認可した AI デバイスの 10% にはすでに PCCP が組み込まれています。しかし、この節約効果は米国に限ったものであり、それがレポート全体の鍵となっている。
さまざまな国で AI を医療機器として登録するにはいくらかかりますか?
上記の政府手数料は請求額の半分にすぎません。残りの半分は専門家の仕事です。つまり、書類の作成、現地登録の保持、当局からのあらゆる質問に対応し、更新とアルゴリズムの変更を提出することです。これは、業界が最も不透明なところです。ほとんどの規制コンサルタント会社は、時間単位で請求するか、各申請を個別に見積もるため、本当のマルチマーケットコストは、変更命令が到着した後に初めて現れます。
Pure Global は、登録ごとに単一の定額の年間料金を公表した最初の医療機器市場アクセス会社です。 から デバイスごと、市場ごと、年間 2,000 米ドル、通常は時間単位で請求されるサービス、つまり国内代理、提出(参照承認に基づく)、更新、変更、および保健当局とのすべての通信が 1 つの料金にまとめられます。タイムシートやメールごとのサプライズはありません。
Pure Global に地域の代表者として活動してもらい、市場ごとに AI 医療機器の登録を行うのにかかる費用は次のとおりです。 (AI SaMD は通常、高リスク クラスに分類されるため、段階的市場には上の数字が適用されます。)
Pure Global 国内代理店 — AI デバイスごとに定額の年間料金
市場ごとに、1 年ごとに 1 つの透明な番号があり、その登録に関するすべてが含まれます。
| 市場 | Pure Global が提供するローカル ロール | 定額年会費 (USD) |
|---|---|---|
| 米国 | FDA US エージェント | $1,000 |
| 欧州連合 | EU 正式な代表者 | $2,000 |
| イギリス | UK 責任者 (UKRP) | $2,000 |
| オーストラリア | TGA スポンサー | $2,000 |
| シンガポール | 登録者 | $2,000 · $3,000 (クラス C/D) |
| マレーシア | 正式な代表者 | $2,000 · $3,000 (クラス C/D) |
| タイ | 正式な代表者 | $2,000 · $3,000 (クラス 3/4) |
| インドネシア | 正式な代表者 | $2,000 |
| ベトナム | 市場認可保有者 | $2,000 |
| 香港 | 現地責任者 | $2,000 · $3,000 (クラス III/IV) |
| マカオ | ライセンス所有者と登録 | $2,000 · $3,000 (クラス III) |
| ブラジル | ブラジル登録保持者 (BRH) | $2,000 · $3,000 (クラス III/IV) |
| メキシコ | メキシコ登録者 | $2,000 · $3,000 (クラス II/III) |
| コロンビア | INVIMA 代表者 | $2,000 · $3,000 (クラス IIb/III) |
1 人の国内代理人に対する定額の年間料金 AI SaMD;これには、参照承認、更新、変更、および当局への対応に関する提出が含まれます。出典: Pure Global マスター価格リスト、2026 年 (登録ごと。複数登録と 3 年契約の割引が適用されます)。
ワンタイム 提出と編集 市場が完全な文書の作成を必要としている場合の研究は、同様に透過的に公開されます。 510(k) コンパイルは $15,000 ~ $20,000 で実行されます、EU 技術文書または CER プロジェクトがリストされています クラス別 ($8,000 – $30,000)、カナダの登録編集物は次のとおりです。 クラスごとに $3,000 ~ $25,000、そして調節経路の決定はフラットです $5,000。すべての数値は時間ごとに引用されるのではなく、事前に引用されます。
実用的な例 — 1 つの AI イメージング アルゴリズム、4 つの市場。 FDA (クラス II) および CE マーク (クラス IIb) によってすでに認可されている単一の AI 放射線診断ツールを、米国、欧州連合、ブラジル、シンガポール全土で 1 年間使用し続けたいとします。 Pure Global の国内代表者の合計 $1,000 (US) + $2,000 (EU) + $3,000 (ブラジル) + $3,000 (シンガポール) = 年間 $9,000 — フラットあらゆる更新、修正、権限交換が含まれます。市場が実際に新しい書類を必要とする場合にのみ、1 回限りの提出作業を追加します。この予測可能性が重要です。市場ごとにルールが異なり、モデルが変わり続ける場合、メーカーが同様の規制法案を提出することは最も避けるべきことです。
収束のパラドックス
ここに明らかな良いニュースがあります。 15 の管轄区域のパッチワークの下で、強力な機構が稼働しています。 調和。 IMDRF は、定義と Good Machine Learning Practice の原則を調整します。の 医療機器単一監査プログラム (MDSAP) 単一の品質システム監査で満足できるようにする 一度に5つのレギュレーター — 米国、カナダ、ブラジル、日本、オーストラリア (FDA)。そして、信頼経路は急速に広がっています。シンガポールは 5 つの照会機関を受け入れ、申請の最大 98% を要約審査を通じてルーティングしています。ブラジル、メキシコ、オーストラリア、マレーシア、ベトナム、湾岸諸国はいずれも外国の承認をある程度認めています。 2026 年 2 月には、IMDRF がグローバルな 信頼性ハンドブック 実践を成文化する(IMDRF N89).
どの市場がどの外国の承認を受け入れるか(依存ルート)
FDA クリアランスまたは CE マークは、数十の市場へのマスター キーであり、それぞれに独自のロックが付いています。
| 市場 | 認知された参考機関/プログラム | 効果 |
|---|---|---|
| シンガポール (HSA) | US FDA、EU NB、カナダ保健省、TGA、日本 厚生労働省 | 要約/迅速化/即時; ~98% が対象 |
| ブラジル (ANVISA) | TGA、カナダ保健省、US FDA、日本 厚生労働省 (クラス III/IV) | 「最適化された分析」最大 20 ~ 30% 高速化 |
| メキシコ (COFEPRIS) | すべての IMDRF メンバー + MDSAP 参加者 | 短縮パスウェイ、30 営業日 |
| オーストラリア (TGA) | US FDA、カナダ保健省、厚生労働省/PMDA、EU NB、MDSAP | 簡易適合性評価 |
| マレーシア (MDA) | US FDA、カナダ保健省、TGA、EU CE、PMDA、HSA、タイ語 FDA | 検証(短縮)ルート+MDSAP |
| ベトナム (MOH) | US FDA、EU、PMDA、TGA、カナダ保健省、MFDS、NMPA | 異常に広範囲にわたる SRA ファストトラック |
| サウジアラビア (SFDA) | FDA/CE サポートのみ | 完全な技術ファイルのレビューが依然として必要です |
| アラブ首長国連邦 (EDE) | CE、US FDA | 信頼性ベースの登録 |
| MDSAP (1 回の監査) | US、カナダ、ブラジル、日本、オーストラリア | 単一の QMS 監査が 5 つ全員によって承認されました |
出典: HSA、ANVISA、COFEPRIS、TGA、MDA マレーシア、タイ語 FDA、ベトナム MOH、SFDA、EDE — Pure Global 分析、2026 年 6 月。
のために 静的 1 つの強力な承認 (通常は FDA または CE) が、大幅なスピードとコストで数十の市場を開くマスターキーになります。これはまさに、うまく運営されている市場アクセスプログラムが活用するために構築されたレバレッジです。
そしてここにパラドックスがあります。 のために アダプティブ AI、マスターキーはまさに最も重要なポイント、つまり変更制御の時点で動作を停止します。収束は次のとおりです デバイス;乖離は上にあります AI。同じ機械学習モデルがアルゴリズムを更新しようとしていると考えてみましょう。
- で 米国、事前に承認された PCCP により、新たに送信せずにアップデートを出荷できます。
- で EU、「大幅な」ソフトウェア変更は依然として認証機関の再審査を引き起こし、その上に別の AI 法適合性評価が重ねられます。
- で 中国、更新は「コアアルゴリズムが変更されていない」場合にのみ許容されます。本物の再トレーニングとは、完全な変更登録を意味します。
- で 韓国、DMPA は事前承認された変更計画を許可しますが、それは事前承認されたパラメーターの範囲内でのみです。
ある分析が率直に言うと、 「FDA によって認可された PCCP は EU AI 法の義務を満たしていません。またその逆も同様です。」 (バークレー ライフサイエンス)。 MDSAP 監査も依存ルートもこれを解決しません。 AI の開発者が 10 の市場で認可を勝ち取ったとしても、平和を買ったわけではありません。彼らは買った 10 種類の変更管理義務、それぞれモデルが改善されるたびにトリガーされます。改良し続けることが価値提案全体であるテクノロジーにとって、それは成功に対する構造的で重なり合う重荷であり、最良のモデルを構築する小規模で動きの速いチームに最も大きな負担がかかります。
次のフロンティア: 生成 AI と基礎モデル
適応的な AI がシステムに負担をかけた場合、生成的な AI はシステムを圧倒する恐れがあります。上記のすべては、出血の検出、駆出率の測定、小結節のフラグなど、単一の明確に定義された使用目的向けにモデルがトレーニングされていることを前提としています。大規模な言語モデルとマルチモーダル基盤モデルは、その前提を 3 つの方法で一度に打ち破ります。 汎用 (1 つのモデルで多くの用途が可能)、 非決定的 (同じプロンプトでも異なる答えが得られる場合があります)、次のような傾向があります。 幻覚 (自信があり、流暢に、間違っています)。これらのプロパティはいずれも、固定された使用目的と「ロックされた」参照バージョンを中心に構築されたフレームワーク内に快適に適合しません。
規制当局はそれを知っています。の 誰が は、2024 年 1 月に大規模なマルチモーダル モデルに特化した初のグローバル ガイダンスを発行し、特に捏造された出力、自動化バイアス、インターネット規模のデータでトレーニングされたシステムの検証の難しさについて警告しました (誰が)。の FDA さん デジタル ヘルス諮問委員会は、2024 年 11 月の初回会議で、生成的な AI 対応デバイスの製品ライフサイクル全体の課題に焦点を当てました (FDA)。そして 韓国特徴的には、最初に、2025 年 1 月に生成医療機器に関する世界初のガイドラインを発行し、2026 年 4 月に最初のそのような機器を認可しました。
しかし、ガイダンスはクリアランス経路と同じではありません。 2026 年半ばの時点で、確立されたルート (510(k)、De Novo、CE マーキング) は依然として、ピンを特定し、固定規格に照らしてテストし、凍結できるデバイスを想定しています。汎用臨床 LLM はこれらの前提条件をまったく満たしません。そのため、「ヘルスケアにおける生成 AI」の第一波は、主に、明確な診断デバイスとしてではなく、デバイス定義を回避する管理ツール (アンビエント スクライブ、文書補助) として市場に到達しました。自律的で生成的な臨床 AI のための規制フロンティアは、正真正銘、まだ描かれており、最初にそれを引き出す市場 (今日の韓国、その後に続く他の市場) が、世界の他の国々がそれを模倣する方法を形作ることになります。開発者にとって実践的な教訓は、どこに境界線が設定されているかを観察し、移動するターゲットに合わせて製品だけでなく規制戦略を設計することです。
市場アクセスの戦略
問題が断片化である場合、その答えはシステムです。上記のパターン全体で、AI 医療機器を世界に送り出し、そこに維持するための反復可能な戦略が生まれます。
1. 証拠を構築する前に分類します。 同じ製品でも、市場とクレームに応じてクラス II またはクラス III になることがあります。ターゲット市場の分類ルールをマッピングする まずなぜなら、それらは必要な臨床的および技術的証拠を決定するからです。主張と証拠を最も厳格なターゲット市場に向けて順序付けすることで、後で関係書類を再構築することを回避できます。
2. 強力なアンカーの承認を獲得し、その依存を意図的に利用します。 FDA クリアランスまたは CE マークは、1 つの市場をはるかに超える価値があります。これは、シンガポール、ブラジル、メキシコ、オーストラリア、湾岸などの短縮ルートのロックを解除するための資格です。芸術とは知ることである どの 各ターゲット市場が認識するアンカーを設定し、それに応じて書類をルーティングします。サウジアラビアは、外国が承認した AI であっても現地での検証を要求していますが、これは信頼が包括的なものではなく地図であることを思い出させます。
3. 国内代表が義務付けられている場所(ほとんどの場所)で立ち上がる。 外国の製造業者は、ブラジル (BRH)、EU (認定代表者)、中国 (法定代理人)、日本 (MAH/DMAH)、サウジアラビア、UAE、インドなどで独自の登録を行うことはできません。それぞれの地域の法人が登録を保持し、保健当局に提出する必要があります。そのような実体を 30 個立ち上げることは非現実的です。単一のパートナーにアウトソーシングすることで、規模を拡大することが可能になります。
4. 変更管理を第一級の複数市場のワークストリームとして扱います。 これは AI 固有の規律です。米国の PCCP を構築するだけでなく、各市場がどのように対応するかをマッピングします。 同じ 更新 — そして、ビジネスにとって重要な最も制限的な体制に合わせてアルゴリズムのリリース リズムを設計します。ライフサイクル計画は現在、市場アクセス計画の一部となっています。
5. 30 個の切断されたファイルではなく、1 つの接続された操作として実行します。 費用は 1 回の登録ではありません。それは調整です。翻訳、現地の所有者、更新カレンダー、変更通知、そして独立して動く数十の体制にわたる市場投入後の警戒です。
実際の典型的なシーケンス。 イメージング-AI 開発者の場合、ロールアウトが頻繁に実行されます。 FDA 510(k) または CEマーク アンカーとして。並行して、銀行の初期収益を得るために、自国市場と信頼性の高い 1 つの市場 (シンガポールまたはオーストラリア) に申請します。これらの承認を使用して、短縮ルートを開きます ブラジル、メキシコ、湾岸;次に、多大な努力と価値の高い市場に挑戦します。 中国と日本 — 現地での検査や臨床データが避けられず、リードタイムが最も長くなる場合。全体を通じて、単一の変更管理計画が集中的に維持され、各市場の更新ルールにマッピングされるため、モデルの改善は必要な場所にファイルされ、必要のない場所にはファイルされません。順序は任意ではありません。キャッシュフロー、証拠の再利用、希少資源市場の順序を意図的に決定します。
これがその作品です Pure Global 国内での代表と、AI が支援する世界中の規制執行を行うために構築されています。 30 以上の市場、業界がデフォルトとしている無期限の時間単位モデルではなく、定額の年間料金で提供されます。このレポートのデータは、FDA、EUDAMED、NMPA、PMDA、MFDS、ANVISA、および数十の国内レジストリと、当社独自の市場ごとのコストおよびタイムラインのデータセットから抽出されたもので、当社がクライアントの世界展開の順序を決定するために使用するインテリジェンスと同じです。これほど徹底的に迷路をマッピングするポイントは、クライアントが迷路を素早く通過できるようにすることです。
結論: 得るべき4つのこと
AI は現在主流のデバイス カテゴリであり、データがそれを証明しています。 2019 年の FDA の認可数はおよそ 700 台に 1 台から、2025 年には 28 台に 1 台にまで増加し、米国だけでも 1,500 台以上の AI デバイスが認可され、韓国、中国、台湾、日本の国家プログラムが急速に拡大しているため、AI SaMD は、目新しいものから標準的なものになりました。
すべての規制当局はそれを上位分類しており、ほとんどはライフサイクル監視に集中しています。 EU およびブラジル、中国のクラス III、EU AI 法に基づく高リスクの規則 11 — 診断および治療 AI は重篤なものとして扱われ、共通の答えは製品ライフサイクル全体の管理、GMLP、および事前に決定された変更計画です。
Reliance は、AI の部分を除いて 1 回の承認トラベルを行います。 調和機構 (IMDRF、MDSAP、依存ルート) により、強力なアンカー承認が真に数十の市場の扉を開くことができます。しかし、adaptive-AI 変更制御は急激に分岐するため、モデルが進化しても単一の承認がどこでも有効であるとは限りません。このギャップは、この分野の運用上の決定的な課題です。
競争力は登録ではなく、登録機にあります。 ルールが市場ごとに異なり、毎年変更される場合、永続的な利点は、どこでも迅速に登録でき、モデルの更新のたびに承認を継続できる、つまり 1 つの接続されたシステムとして維持できる人にあります。
ご相談ください
AI 医療機器を構築または拡張し、どの市場にどのような順序で参入し、モデルの改善に合わせて各承認を有効に保つかを検討している場合、それがまさに私たちが解決する問題です。 Pure Global に話しかけてください 上記のデータに基づいて構築された市場アクセス計画について — または、 市場ごとの登録ガイド 単一の国についてさらに深く掘り下げることができます。
情報源
上でインラインで引用した政府および調和団体。主要な参考文献を以下にグループ化します。すべての数字には日付が付けられています。市場規模と予測数値は第三者による推定であり、その範囲はさまざまであり、範囲として読み取る必要があります。
定義、フレームワーク、基本原則
- IMDRF — SaMD: キーの定義 (N10、2013); リスクの分類 (N12、2014); ML 対応医療機器: 重要な用語 (N67、2022); GMLP の指導原則 (N88、2025); 信頼性ハンドブック (N89、2026)。 imdrf.org
- US FDA — 医療機器としてのソフトウェア (SaMD); AI/ML ベースの SaMD に対する修正のための規制枠組みの提案 (2019); AI/ML アクション プラン (2021年)。 fda.gov
- 誰 — AI の健康のための倫理とガバナンス (2021); AI に関する健康に関する規制上の考慮事項 (2023); LMM ガイダンス (2024); 適切な信頼性の実践、TRS 1033 付属書 10 (2021)。誰.int
米国
- FDA — 人工知能対応医療機器 リスト (2026 年第 1 四半期更新、1,524 台のデバイス、約 76% が放射線学)。 PCCP 最終ガイダンス (2024 年 12 月); デジタルヘルス諮問委員会 生成的な AI (2024 年 11 月); MDUFA 2026年度手数料。 fda.gov · The Imaging Wire (放射線学シェア分析、2026 年)。 Innolitics および IntuitionLabs (クリアランス トラッカー、2025 ~ 26 年)。
欧州連合と UK
- EUR-Lex — 規則 (EU) 2017/745 (MDR)、附属書 VIII 規則 11;規則 (EU) 2024/1689 (AI 法)、第 6 条、99 条、113 条。欧州委員会 — MDCG 2019-11 Rev.1 および MDCG 2025-6。 MedTech ヨーロッパおよびチーム NB (公認機関の能力)。 MHRA — ソフトウェアおよび AI 変更プログラム。 AI エアロック。 CE-認識ガイダンス。英国政府
カナダ、オーストラリア、日本、韓国
- カナダ保健省 — ML 対応医療機器の市販前ガイダンス (2026 年 4 月)。カナダ.ca
- TGA — ソフトウェア改革 (2021); AI の協議結果 (2024 ~ 2026 年)。 tga.gov.au
- PMDA/MHLW — SaMD ガイダンス; SaMD の場合はダッシュ。いだてん。 pmda.go.jp
- MFDS — 2025 承認レポート (153 AI デバイス); DMPA;生成-AI ガイドライン。 mfds.go.kr; bioin.or.kr
アジア太平洋、中南米、MEA
- HSA シンガポール — GL-04-R4;信頼/参照機関。 hsa.gov.sg; moh.gov.sg
- NMPA 中国 — AI 分類と 2025 年 10 月改革。 nmpa.gov.cn · JMIR 医療情報学 (154 AIMD、2026 年)。
- CDSCO インド — MD ソフトウェア ガイダンス草案 (2025 年 10 月)。 TFDA 台湾 — CADe/CADx ガイダンス。 J. フォルモス。医学。准教授(166 ライセンス)。
- ANVISA ブラジル — RDC 751/2022 (規則 11)、RDC 657/2022、IN 290/2024。 gov.br/アンビザ
- COFEPRIS メキシコ — 略称パスウェイ (2025)。 SFDA サウジアラビア — MDS-G010 (2022)。 UAE EDE — 連邦政令法 38/2024。 SAHPRA 南アフリカ — AI 通信 (2025)。
市場、企業、特許、安全性
- 市場規模: Grand View Research;マーケットサンドマーケット;ビジネスリサーチ会社。モルドールのインテリジェンス。フォーチュン ビジネス インサイト (2025 ~ 26 年)。
- 資金と企業: Rock Health (2025 年のデジタルヘルス資金調達)。 CBインサイト;会社の 2025 年度提出書類 (Tempus AI、iRhythm、HeartFlow、Butterfly Network、Lunit、VUNO)。アイドック(シリーズE)。
- 特許: WIPO Technology Trends (2019) & Generative AI Patent Landscape (2024);クニパ。
- 安全性: NEJM (パルスオキシメーターバイアス、2020); JAMA 内科 (エピック敗血症モデル、2021); JAMA ヘルス フォーラムおよび JAMA ネットワーク オープン (AI を思い出す、2025 年); FDA パルスオキシメーターのガイダンス。
- 貿易: OECD-IMF-WTO デジタル取引の測定 (2021年); WTO;アンタッド。
- Pure Global 独自の市場アクセスコストとタイムラインデータセット (2026 年)。 openFDA、EUDAMED および MFDS データベース分析 — Pure Global、2026 年 6 月。



どこにいても、
ご相談ください。
詳しい情報が必要な段階でも、協業を始める段階でも、規制対応の各ステップをサポートします。
お問い合わせ