
L'IA en tant qu'instrument médical: la carte mondiale de la réglementation, de l'enregistrement et de l'accès au marché
L'intelligence artificielle est la classe d'instruments médicaux qui connaît la croissance la plus rapide dans l'histoire, et la plus diversifiée en matière de réglementation. La FDA des États-Unis énumère maintenant 1 524 dispositifs compatibles avec l'IA; la Corée du Sud a autorisé 153 en une seule année. Pourtant, le même logiciel est la classe II aux États-Unis, la classe IIa+ et «à haut risque» dans l'UE, et la classe III en Chine — chacun avec ses propres preuves, les détenteurs locaux et les règles de contrôle du changement. Ce rapport montre comment chaque organisme de réglementation majeur classe, approuve et police l'IA comme un instrument médical, avec des coûts d'enregistrement, des délais et des itinéraires de dépendance sur plus de 30 marchés, et le playbook pour les atteindre sans reconstruire le dossier chaque fois.
Un guide de terrain basé sur des données probantes sur la classification, l'approbation et la police de l'intelligence artificielle dans le domaine des soins de santé et sur la façon d'atteindre plus de 30 marchés sans reconstruire le dossier à chaque fois.
TL;DR
L'intelligence artificielle est devenue la catégorie de dispositifs médicaux qui connaît la croissance la plus rapide de l'histoire. La liste publique des dispositifs à AI de la Food and Drug Administration des États-Unis a atteint 1 524 autorisations au milieu de 2026, avec environ les trois quarts en radiologie (FDA). Selon notre propre analyse de la base de données FDA 510(k), la part d'IA de toutes les autorisations est passée d'environ 1 en 700 en 2019 à environ 1 en 28 en 2025. La Corée du Sud seule a autorisé 153 dispositifs AI en 2025 (MFDS).
Mais le même logiciel qui navigue à travers une agence peut s'arrêter dans le prochain. Voici l'argument de ce rapport, en cinq points:
- L'AI est un dispositif, et la plupart d'entre eux sont des « logiciels comme instrument médical » (SaMD Lorsque le logiciel diagnostique, trie ou recommande un traitement, il est régulé comme un scalpel ou un scanner - dans des dizaines de juridictions, chacune ayant ses propres règles.
- Le monde entier le classe. La règle 11 de l'UE relative aux MDR, la règle 11 du Brésil, le défaut de la Chine de la classe III pour le diagnostic de l'IA — logiciel diagnostique et thérapeutique se situe presque partout dans une classe à risque élevé qui exige un examen par une tierce partie.
- L'IA a cassé le modèle d'approbation unique. Un modèle qui continue d'apprendre les changements après qu'il soit livré, de sorte que les régulateurs ont inventé de nouveaux mécanismes — le plan de contrôle du changement préétabli des États-Unis, l'IDATEN du Japon, la DMPA de la Corée — qui ne se correspondent pas .
- La confiance converge, l'IA diverge. Un réseau de voies de dépendance et de reconnaissance est censé permettre à une approbation de débloquer de nombreux marchés. Il fonctionne — pour les appareils statiques. Dans le cas de l'IA adaptée, "un PCCP autorisé par la FDA ne satisfait pas aux obligations de la loi sur l'IA de l'UE, et vice versa" (Berkley Sciences de la vie).
- Les gagnants industrialisent donc l'enregistrement multi-marchés. L'avantage concurrentiel n'est plus une simple autorisation; c'est la machine opérationnelle qui transforme une approbation en trente, et maintient chacun valide à mesure que l'algorithme évolue.
Voilà l'écart.Pure Global clôture — représentation dans le pays et exécution réglementaire assistée par AI sur plus de 30 marchés, moyennant une redevance annuelle forfaitaire. Le reste de ce rapport est la carte.
Qu'est-ce que l'AI en tant qu'instrument médical?
Commencez par le mot qui fait le levage lourd: dispositif. Si un logiciel est « destiné à être utilisé dans le diagnostic, le traitement ou la prévention de la maladie », il s'agit d'un dispositif médical dans presque tous les systèmes juridiques du monde — qu'il fonctionne sur une puce à l'intérieur d'un scanner IRM ou comme une application dans le navigateur d'un radiologue. L'IA n'obtient pas son propre statut dans la plupart des pays; elle hérite de tout l'appareil de la loi sur les dispositifs médicaux.
La définition d'ancrage provient du International Medical Device Regulators Forum (IMDRF), l'organisme où les grandes agences harmonisent leur vocabulaire. Dans son document de base de 2013, IMDRF a défini Logiciel comme un dispositif médical (SaMD) comme "logiciel destiné à être utilisé à une ou plusieurs fins médicales qui accomplissent ces fins sans faire partie d'un dispositif médical matériel" (IMDRF N10). La FDA a adopté ce langage mot pour mot et établit une distinction à trois sens qui compte pour tout en aval (FDA):
- SaMD — un logiciel qui est le dispositif médical (un algorithme de triage thorax-X, un détecteur de rétinopathie diabétique). C'est là que vit le plus d'IA clinique.
- SiMD — logiciel dans un dispositif médical, intégré au matériel (le micrologiciel utilisant une pompe à perfusion).
- Logiciel utilisé pour fabriquer ou entretenir un appareil, qui est régulé différemment.
Pour l'IA en particulier, le document de 2022 de l'IMDRF définit un instrument médical à apprentissage automatique comme un instrument médical qui utilise l'apprentissage automatique, en partie ou en totalité, pour atteindre l'objectif médical visé.IMDRF N67).
La distinction qui casse tout: verrouillée contre adaptative
La réglementation des appareils traditionnels repose sur une simple affaire: vous prouvez qu'un appareil est sûr et efficace une fois, à une conception fixe, et que la conception reste mise. L'IA brise cette hypothèse. Le document de travail pivot 2019 de la FDA a tracé la ligne avec précision. Un algorithme "verrouillé" est celui qui "fournit le même résultat chaque fois que la même entrée lui est appliquée et ne change pas avec l'utilisation" — une table de recherche, un arbre de décision, un classificateur gelé (FDA, 2019). Un algorithme adaptatif ou d'apprentissage continu change après son déploiement.
Cette propriété unique, un logiciel qui s'améliore sur le terrain, est la raison pour laquelle l'IA a besoin d'une décennie de nouvelle réglementation. Si le produit que vous avez approuvé en janvier n'est pas le produit qui circule dans un hôpital en juin, qu'avez-vous approuvé exactement? Chaque cadre dans le présent rapport est, au fond, une tentative de répondre à cette question, et le cycle de vie du produit total (TPLC) — surveillance tout au long de la vie de l'appareil, pas seulement le moment de l'autorisation — est la réponse partagée.
Comment les régulateurs décident de la difficulté à regarder
Le cadre de classification des risques de 2014 de l'IMDRF définit la logique que le monde suit maintenant:SaMD mérite des échelles avec deux facteurs — l'importance de l'information qu'elle fournit (est-ce que c'est information, drive, ou diagnose/traitement?) et la gravité de la situation en matière de soins de santé (non grave, grave ou critique) (IMDRF N12). Une application qui suggère des étirements pour la douleur dorsale et un algorithme qui signale une saignement du cerveau ne sont pas le même animal réglementaire, et cette grille à deux axes est pourquoi.
L'Organisation mondiale de la santé a ajouté l'échafaudage éthique. Son rapport 2021 L'éthique et la gouvernance de l'intelligence artificielle pour la santé ont fixé six principes: protéger l'autonomie; promouvoir le bien-être et la sécurité; assurer la transparence et l'explicabilité; promouvoir la responsabilité et la responsabilisation; assurer l'inclusion et l'équité; et promouvoir l'IA réceptive et durable (WHO). L'OMS a suivi avec des considérations réglementaires en 2023 et, en janvier 2024, les premières orientations mondiales visant carrément à générer de l'IA et de grands modèles multimodal (WHO).
Comment nous sommes arrivés ici
Les dispositifs médicaux de l'IA ne sont pas arrivés avec une seule percée; ils ont accrété, pays par pays, sur environ une décennie. Les jalons ci-dessous retracent comment une définition en 2013 est devenue des règles dédiées du cycle de vie sur cinq continents.
Un calendrier global de la réglementation des dispositifs médicaux de l'IA (2013-2026)
En à peine une décennie, AI SaMD est passé d'une définition IMDRF à des règles de cycle de vie dédiées sur cinq continents.
| Date | Étape |
|---|---|
| Déc 2013 | L'IMDRF N10 définit le « logiciel comme instrument médical » (SaMD) |
| Janvier 2017 | La FDA élimine les artères — premier nuage + outil clinique d'apprentissage profond (510k) |
| Avr 2018 | La FDA autorise IDx-DR — premier diagnostic autonome de l'IA (De Novo) |
| Mai 2018 | Corée MFDS approuve VUNO Med-BoneAge — Le premier appareil AI coréen |
| Déc. 2018 | Japon PMDA approuve EndoBRAIN (classe III) — la première AI du Japon SaMD |
| Avr 2019 | Document de travail de la FDA sur les modifications à apporter à l'IA/ML SaMD(verrouillé par rapport à adaptatif) |
| 2020 | Chine NMPA approuve DeepVessel FFR — premier appareil AI de classe III; le Japon lance IDATEN + DASH pour SaMD |
| Janv. 2021 | FDA AI/ML Plan d'action; juin 2021 OMS six principes éthiques |
| Oct 2021 | GMLP — 10 principes directeurs (FDA + Santé Canada + MHRA du Royaume-Uni) |
| 2022 | Saudi SFDA MDS-G010 — première ligne d'orientation dédiée aux dispositifs d'IA (citée par certains comme étant la première « en vigueur »); termes IMDRF N67 ML; Brésil RDC 751/657 |
| Août 2024 | La loi de l'UE sur l'IA entre en vigueur; les directives de l'OMS sur les MMT (generative-AI) (janvier 2024) |
| Déc 2024 | La FDA met la dernière main aux lignes directrices du Plan de contrôle du changement (PCCP) prédéterminé |
| Janv. 2025 | IMDRF N88 GMLP final; la Corée délivre la première directive mondiale sur les dispositifs générateurs d'IA; FDA AI cycle de vie projet de guide |
| Fév 2026 | IMDRF N89 Livre de lecture sur la fiabilité |
| Avr 2026 | Santé Canada met la dernière main aux lignes directrices sur les appareils ML; la Corée approuve le premier appareil generative-AI |
Source: Recueil de documents primaires de l'IMDRF, de la FDA des États-Unis, de l'UE, de la NMPA, de la PMDA, de la MFDS, de la SFDA, de l'ANVISA, de Santé Canada et de l'OMS —Pure Global, juin 2026
Quelques instants méritent d'être soulignés. En janvier 2017, la FDA a éliminé les artères — le premier outil clinique à combiner l'informatique en nuage et l'apprentissage profond (RP Newswire). Puis, en avril 2018, est venu le bassin versant: la FDA a autorisé IDx-DR, la première IA permis de donner un diagnostic autonomement, sans médecin interprétant le résultat — un dépistage diabétique-rétinopathie pour les soins primaires qui ont atteint 87,2% sensibilité et 90,7% spécificité dans son essai pivot (npj Médecine numérique). En quelques mois, la Corée (VUNO Med-BoneAge, mai 2018) et le Japon (EndoBRAIN, décembre 2018) ont approuvé leurs propres premières; La Chine a suivi en 2020 avec DeepVessel FFR, son premier appareil AI de classe III (npj Médecine numérique).
L'échafaudage réglementaire s'est arrêté entre 2021 et 2025: les principes trilatéraux Bonne pratique d'apprentissage automatique en octobre 2021, la Loi sur l'AI de l'UE qui entre en vigueur en août 2024, les directives finales de la FDA Plan de contrôle du changement préétabli en décembre 2024, et — vous dire à quelle vitesse la frontière se déplace — la ligne directrice de la Corée sur les dispositifs générateurs d'AI en janvier 2025 (Biomonde). En février 2026, l'IMDRF avait publié un livre de lecture mondial pour aider les régulateurs à s'appuyer mutuellement sur leurs travaux (IMDRF N89).
Les données: quelle taille, quelle vitesse, où
La courbe de clairance fléchit fortement vers le haut
La meilleure mesure de la pénétration de l'IA dans la médecine est le public de la FDA La liste des instruments médicaux à base d'intelligence artificielle, qui a atteint 1 524 autorisations lors de sa dernière mise à jour (données reflétant le premier trimestre de 2026) (FDA). Pour voir la vitesse, nous avons analysé directement la base de données FDA 510(k) sous-jacente. Les clairances identifiées de l'IA/ML sont passées d'une poignée à la fin des années 2010 à 115 en 2025, et, de façon plus éloquente, elles sont passées de 0,14% des clairances de 510k) en 2019 à 3,57% en 2025, soit environ 25 fois plus qu'en six ans.
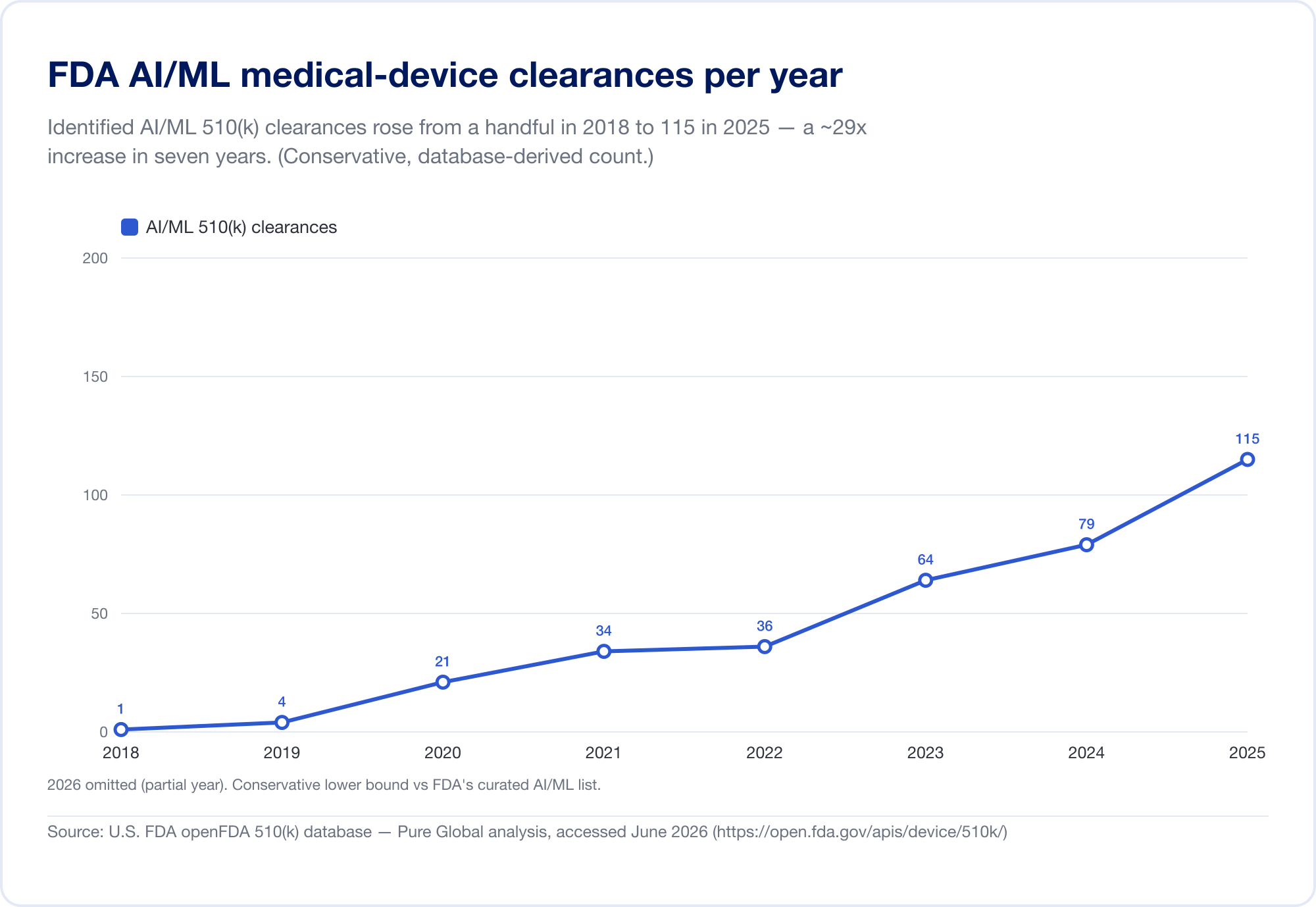
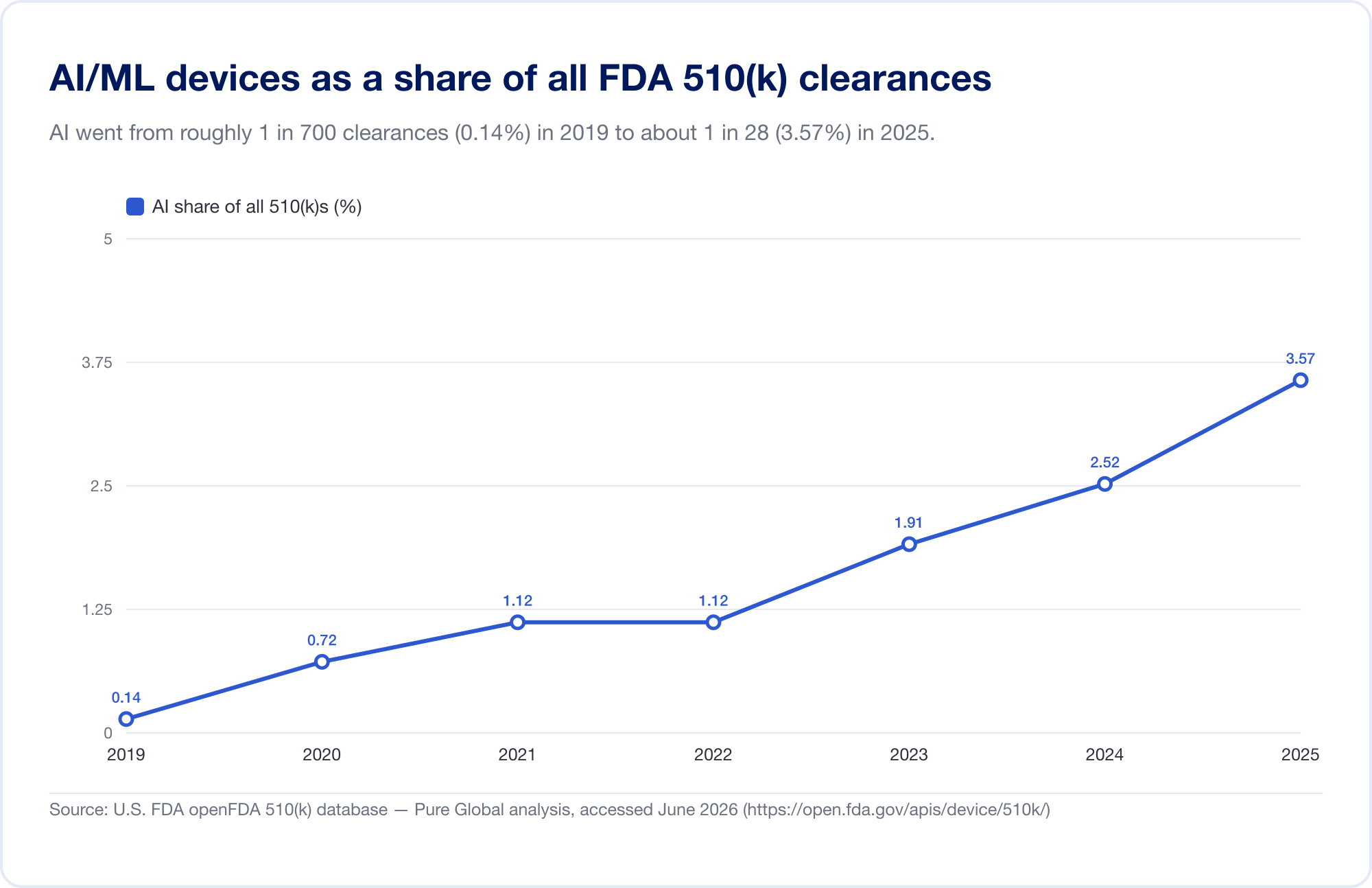
(Notre compte de base de données est délibérément conservateur — plus étroit que la liste de la FDA, parce que beaucoup d'outils de radiologie de l'IA s'assoient sous des codes de produits dont le texte ne dit jamais «AI». Nous l'utilisons pour la tendance et la géographie; la liste de la FDA est le total global. Source: base de données open FDA 510(k) —Pure Global analyse, juin 2026.
C'est en grande partie une histoire d'imagerie — pour l'instant
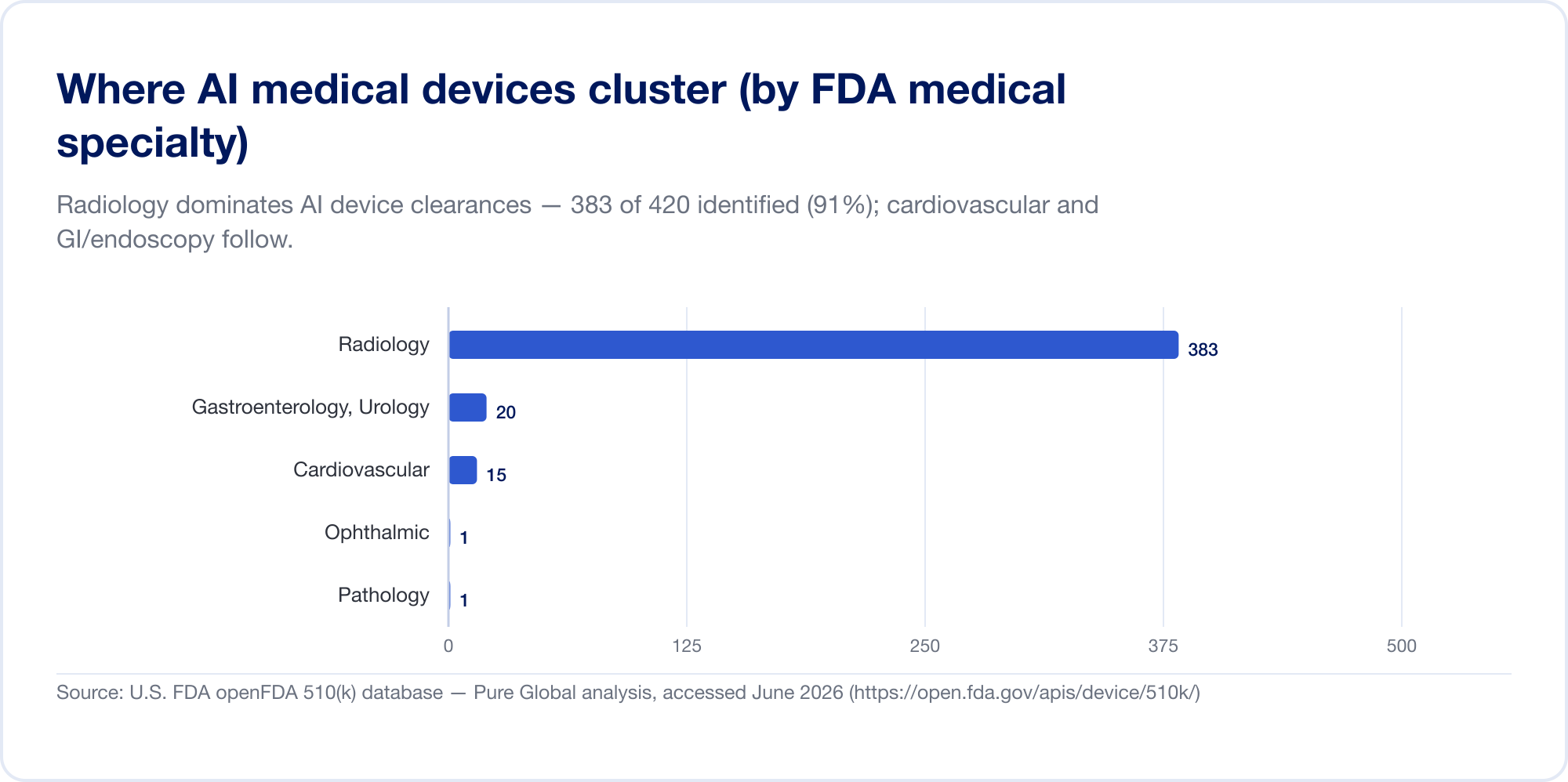
L'IA en médecine est, aujourd'hui, principalement en radiologie. Sur la liste de la FDA, la radiologie représente environ 76% de toutes les autorisations d'IA (Le fil d'imagerie); dans notre propre échantillon de clairance, la concentration est encore plus élevée, avec une cardiovasculaire et gastroentérologie (endoscopie) une distance seconde et troisième. La raison en est structurelle: l'imagerie est numérique, abondante et étiquetée, et la voie 510(k) permet à un nouvel algorithme de citer un algorithme existant comme prédicat. La pathologie, les signaux de cardiologie et les modèles de texte clinique se développent, mais le centre de gravité reste l'image.
Les innovateurs sont partout; les marchés sont partout ailleurs
Voici le fait que devrait recadrer tout plan de commercialisation. Les dispositifs médicaux d'IA dédouanés aux États-Unis proviennent d'au moins 16 pays. Dans notre analyse des demandes d'AI 510(k), les États-Unis dirigent, mais la Corée du Sud, Israël, le Canada, la France, la Chine, l'Allemagne, le Royaume-Uni, les Pays-Bas, Taiwan et le Japon, chaque domaine est significatif.
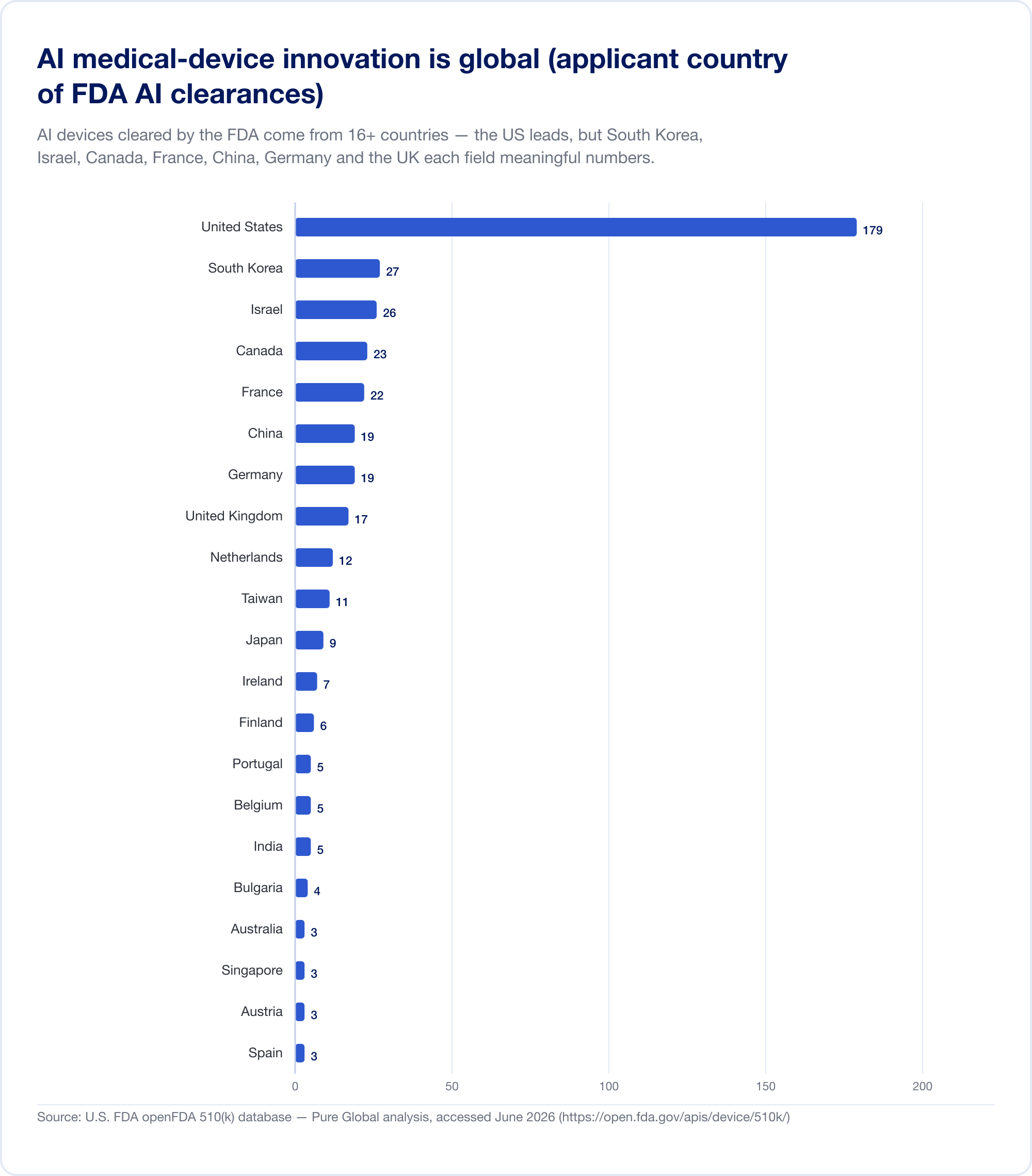
La production nationale confirme la tendance. La Corée du Sud a autorisé (approbations et certifications combinées) 64 dispositifs d'IA en 2023, 108 en 2024 et 153 en 2025 – un bond de 41,6 %, avec 77,7 % de fabrication nationale (MFDS La Chine avait approuvé environ 154 dispositifs médicaux d'IA au milieu de 2025, dont environ 80 % dans la classe III à risque le plus élevé (JMIR Informatique médicale Taiwan a autorisé 166 dispositifs AI/ML entre 2020 et 2024 (J. Formos. Med. Assoc. Le Japon comptait 51 SaMD sur la liste du PMDA en septembre 2025 (Santé mondiale et médecine).
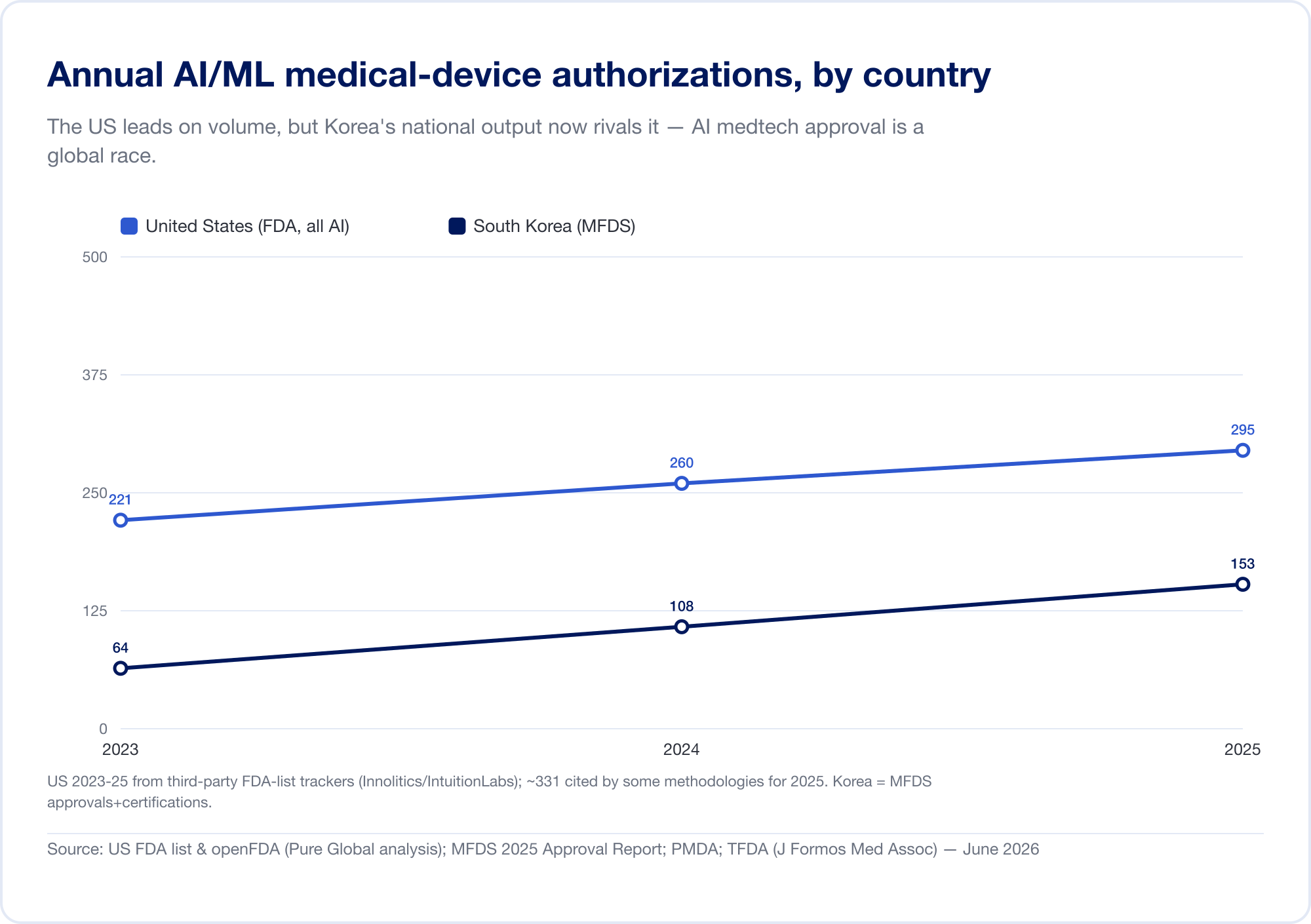
L'implication est directe: un algorithme brillant construit à Tel Aviv, Séoul ou Shanghai doit atteindre les patients sur des marchés où chacun parle un langage réglementaire différent. L'innovation est globale; l'approbation est obstinément locale.
Quelle est la taille du marché? Tout dépend de ce que vous comptez.
Les estimations de la taille du marché pour l'IA dans les soins de santé s'étendent sur un ordre de grandeur, parce que les analystes dessinent la frontière dans des endroits sauvagement différents. La grande catégorie "AI dans les soins de santé" — qui regroupe la découverte de médicaments, l'automatisation administrative, les scribes ambiants, et plus encore — est prévue à 187,7 milliards USD d'ici 2030 à un TCAC de 38,5% (Recherche Grand Vue), avec la maison la plus agressive projetant plus de 1 billion de USD en 2034 (Fortune Business Insights). La tranche plus étroite, réglementée — AI dans les dispositifs médicaux — est beaucoup plus petite et plus crédible: environ 12,4 milliards USD en 2025, atteignant 42,4 milliards USD d'ici 2030 (Société de recherche en affaires) Pure SaMD est estimé à environ 5 à 25 milliards d'USD selon la source (Mordor Intelligence). Traitez chacun de ceux-ci comme une gamme, pas un fait.
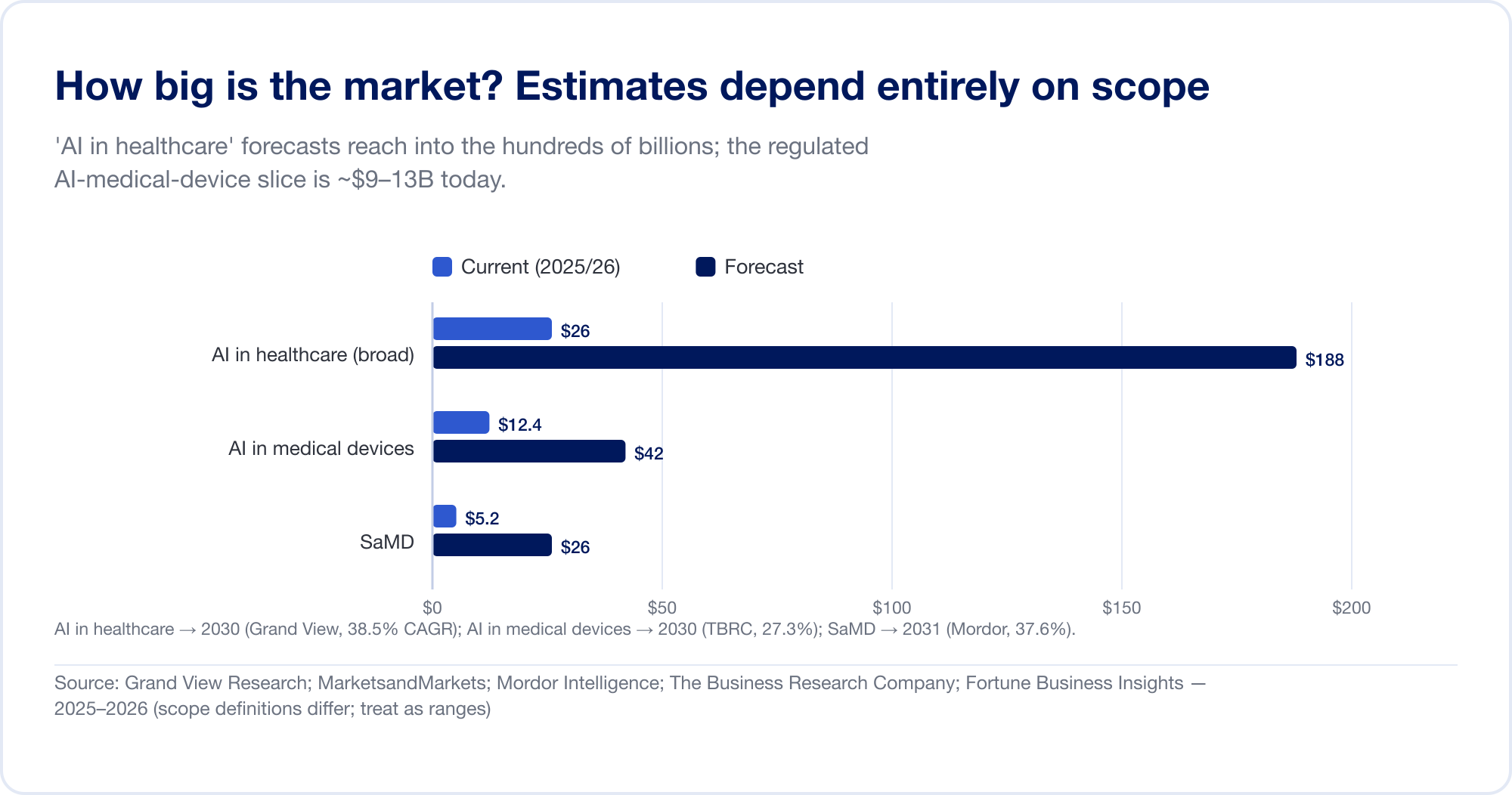
La capitale raconte une histoire plus propre. Le financement des entreprises de santé numérique aux États-Unis a rebondi à 14,2 milliards USD en 2025 (+35 %), et pour la première fois, les start-ups utilisant l'IA ont remporté la majorité — 54 % de tous les dollars (Santé des roches).SaMD de la croissance à la rentabilité: Tempus AI a affiché un chiffre d'affaires de 1,3 milliard USD (+83 %) Tempus); iRhythm a atteint USD 747 millions et son premier trimestre rentable GAAP (iRhythme); HeartFlow est devenu public en août 2025 et a augmenté de 40 % pour atteindre 176 millions de dollars (Coeur); la Corée Lunit a augmenté de 53 % avec 92 % des recettes provenant de l'étranger (Unité). Parmi les joueurs privés, Aidoc a recueilli une série E de 150 millions de dollars en avril 2026, prenant le financement total d'un demi-milliard de dollars (Aidoc).
La course aux brevets a un leader différent
La propriété intellectuelle est là où le concours mondial est le plus visible. L'étude historique de l'OMPI a compté environ 340 000 demandes de brevet liées à l'IA, le domaine des sciences de la vie et des sciences médicales étant le troisième plus important (WIPO). Mais depuis, la géographie s'est effondrée: dans la décennie à 2023, la Chine a déposé 38 210 inventions génératrices-AI — plus que le reste du monde combinés — contre 6 276 des États-Unis (WIPO). Le dernier jour de 2024, l'office chinois des brevets a publié des lignes directrices spécifiques sur l'examen des brevets de l'IA (CNIPA). Le pipeline de l'IA médicale est rempli de façon disproportionnée en Asie, ce qui fait de la stratégie d'accès multimarché une question de quand, pas si, pour une part croissante des développeurs du monde.
L'angle mort: vous ne voyez pas SaMD des données commerciales
Une mise en garde analytique que recadre la façon de penser à ce marché. L'information traditionnelle sur les marchés de la medtech s'appuie sur les données relatives aux importations et aux douanes — mais pure SaMD est invisible à elle. Les logiciels livrés par téléchargement en nuage ou par app store ne traversent aucune frontière physique, ne génèrent pas de déclaration en douane du code SH et ne sont pas saisis dans les statistiques du commerce des marchandises. "Les flux de données qui ne sont pas directement monétisés ne sont généralement pas considérés comme des flux commerciaux dans les normes statistiques actuelles"* (OCDE-FMI-OMC), et le moratoire de l'OMC sur les droits de douane pour les transmissions électroniques a été décrété depuis 1998 (UNCTAD). Le matériel intégré à l'IA* — un scanner CT compatible avec l'IA — se déplace comme un bon et apparaît dans les données; un algorithme nuageux de 510k) ne l'est pas. Le point de départ: analyse commerciale conventionnelle systématiquement sous-comptes logiciels d'IA et matériel surpoids. La seule empreinte fiable d'un SaMD'la diffusion globale est ses enregistrements - qui est exactement la lentille que ce rapport utilise.
Le bilan de sécurité
Derrière chaque resserrement des règles se trouve un ensemble de preuves que l'IA en médecine peut échouer de la manière dont les dispositifs traditionnels ne le font pas. Trois constatations, en particulier, ont modifié la façon dont les organismes de réglementation pensent.
Bais qui se cachent dans des données claires. Une étude historique New England Journal of Medicine a révélé que les oxymètres de pouls — omniprésents et de plus en plus associés à des algorithmes — manquaient de niveaux dangereusement bas d'oxygène dans le sang (hypoxémie occulte) chez 11,7 % des patients noirs par rapport à 3,6 % des patients blancs, ce qui représente une disparité d'environ trois fois plus grande dans environ 50 000 lectures appariées (NEJM, 2020). La FDA a publié une communication sur l'innocuité en février 2021 et, d'ici janvier 2025, un projet d'orientation exigeant une validation plus diversifiée des tons cutanés (FDA). La leçon généralisée: un modèle formé sur une population non représentative peut porter ce biais silencieusement dans chaque hôpital qui le déploie.
Validation qui ne survit pas au contact avec la réalité. Le modèle Epic Sepsis, un outil de prédiction exclusif déployé dans des centaines d'hôpitaux américains, a été validé à l'extérieur lors de 38 455 hospitalisations et a marqué une zone sous la courbe de 0,63, bien au-dessous de la demande du fournisseur de 0,76 à 0,83. Il a manqué 67% des cas de septicémie pendant que le tir alerte sur 18% de tous les patients (JAMA Médecine interne, 2021 Un modèle peut être "en production" à l'échelle et ne fonctionne toujours pas comme annoncé.
Rappelle ce groupe juste après l'autorisation. Une analyse Johns Hopkins/Yale de 2025 des dispositifs d'IA autorisés par la FDA a révélé que 43,4% des rappels d'appareils d'IA ont eu lieu au cours des 12 premiers mois suivant la clairance, soit environ le double du taux global pour 510(k) dispositifs (Forum sur la santé de la JAMA, 2025). Une étude parallèle a révélé que les rappels étaient concentrés parmi les dispositifs qui n'avaient pas fait l'objet d'études cliniques publiées (Réseau JAMA ouvert, 2025 Le contexte est important: environ 97 % des dispositifs AI/ML sont transparents par la voie 510(k), ce qui ne nécessite aucun test humain prospectif, ce qui entraîne une grande vigilance après la mise en marché.
Le calcul de la sécurité: pourquoi les régulateurs se resserrent sur l'IA
Le passage à la surveillance du cycle de vie est attesté par des lacunes dans la validation et des rappels précoces.
| Constatation | Graphique | Source |
|---|---|---|
| Hypoxémie occulte de l' oxyde d' pulse, patients noirs et patients blancs | 11,7 % vs 3,6 % (~3x) | NEJM, décembre 2020 |
| ASC externe du modèle Epic Sepsis (vs 0.76–0.83) | 0,63; oublié 67% des cas de septicémie | JAMA Stagiaire. Med., juin 2021 |
| Rappel des dispositifs anti-IA dans les 12 mois suivant l'autorisation | 43,4 % (~2x toutes les 510k) | Forum sur la santé de la JAMA, 2025 |
| Rappel des dispositifs d'IA (903 études) | 4,8%, concentrés dans ceux qui n'ont pas d'études cliniques | Réseau JAMA ouvert, 2025 |
| Dispositifs d'IA nettoyés au moyen du paragraphe 510(k) (aucun essai potentiel n'est requis) | ~97% | Analyses de la liste de la FDA, 2025 |
Ajoutez à cela le problème du changement de données — les modèles déployés se dégradent silencieusement au fur et à mesure que la population de patients, les scanners ou les systèmes de codage autour d'eux changent — et vous avez la raison d'être de l'ensemble de l'appareil moderne: Bonne pratique d'apprentissage automatique, Plans de contrôle du changement prédéterminés et surveillance obligatoire des performances réelles. Les régulateurs ne se serrent pas parce que l'IA ne fonctionne pas. Ils se resserrent parce que ça marche jusqu'à ce que ça marche tranquillement.
La carte réglementaire mondiale
C'est le noyau de référence du rapport: comment les principales juridictions classifient, examinent et policent réellement l'IA SaMD au milieu de 2026. La ligne passante à regarder est classification (qui classe le risque dans le logiciel) et contrôle de changement (ce qui se passe lorsque l'algorithme est mis à jour). Une matrice de comparaison consolidée suit le détail régional.
Comment 15 juridictions réglementent l'IA en tant qu'instrument médical (2026)
- Même logiciel, quinze réponses: classification, orientations spécifiques à l'IA et règles de contrôle du changement divergent le marché.*
| Compétence | Où la plupart des IA SaMD terres | AI/SaMD des directives | Mécanisme de contrôle du changement/AI adaptatif | Référence à l'agrément étranger |
|---|---|---|---|---|
| États-Unis (FDA) | Classe II (510 k)/De Novo) | Oui — PCCP final 2024; projet de cycle de vie 2025 | PCCP (préautorisation des modifications, aucune nouvelle présentation) | Non (réexamen individuel fondé sur les prédicats) |
| Union européenne | Classe IIa+ (règle 11 du RIM) + Loi sur l'IA à risque élevé | MDR + Loi sur l'IA + MDCG 2025- 6 | Changement important →Notified Body révision + Loi sur l'IA | No (évaluation de la conformité CE) |
| Royaume-Uni (MHRA) | Catégorie IIa+ (RMR du Royaume-Uni 2002) | Logiciel & Programme de changement d'IA; Airlock AI | PCCP prévu (instrument statutaire) | CE accepté en GB à 2028/2030 |
| Canada (Santé Canada) | Classe II–IV | Oui — Lignes directrices ML final avril 2026 | PCCP | MDSAP pour le SGQ; pas la pleine dépendance du produit |
| Australie (TGA) | Catégorie IIa– III | Examen d'IA 2024 (14 constatations) | En cours de développement | Voie de régulation comparable-outre-mer |
| Japon (PMDA/MHLW) | Classe II–III | DASH pour SaMD;SaMD des directives | IDATEN (PACMP) modifications pré-approuvées | Données cliniques étrangères acceptées; PASMD |
| Corée du Sud (MFDS) | Classe 2–3 | Oui — y compris la première ligne directrice mondiale de l'AI | Plans de changement préapprouvés par la DMPA | Limité; propre réexamen |
| Singapour (HSA) | Catégorie B–D | Oui — GL-04-R4 (2025), cycle de vie de l'IA-MD | Notification de changement | Oui — 5 agences de référence (~98% abrégé) |
| Chine (NMPA) | Classe III (logiciel de décision) | Oui — Principes CMDE AI + catalogue de classification | Découper seulement si l'algorithme de base est inchangé | Non (agent dans le pays; essais de type) |
| Inde (CDSCO) | Classe A–D | Projet seulement (octobre 2025) | Protocole de modification de l'algorithme (proposé) | Classe C/D |
| Taïwan (TFDA) | Classe II–III | Oui — CAO/CADx + directives de rédaction du PCCP | Lignes directrices du PCCP (2024) | L'évaluation locale des résultats a été mise en évidence |
| Brésil (ANVISA) | Classe II–IV (règle 11) | CDR 657 (SaMD); aucune règle d'IA spécifique | Enregistrement complet du changement | Oui — IN 290/2024 (classe III/IV, 4 agences) |
| Mexique (COFEPRIS) | Classe I–III | Règles générales relatives aux dispositifs | Réenregistrement | Oui — voie abrégée (IMDRF + MDSAP) |
| Arabie saoudite (SFDA) | Classe A–D | Oui — MDS-G010 (début; cité comme premier «exécutable») | Modification de la notification par le biais du GHAD | Support uniquement; validation locale requise |
| EAU (EDE) | Classe I–III | Règles générales relatives aux dispositifs | Réenregistrement | Oui — reconnaît CE/FDA |
Source: FDA des États-Unis, Loi sur les RMM/AI de l'UE, MHRA, Santé Canada, TGA, PMDA/MHLW, MFDS, HSA, NMPA, CDSCO, TFDA, ANVISA, COFEPRIS, SFDA, EDE —Pure Global analyse, juin 2026.
États-Unis — l'indice de référence, et les plus occupés
Les États-Unis administrent le régime de dispositifs d'IA le plus actif au monde, administré par le Centre for Devices and Radiological Health de la FDA et son Centre d'excellence en santé numérique. Il n'existe pas de « statut d'AI » spécial; les fonctions d'IA qui répondent à la définition de l'instrument sont réglementées comme suit:SaMD par trois voies: 510(k) clairance (montrant « l'équivalence substantielle » avec un dispositif prédictif), Classification De Novo (pour les nouveaux dispositifs à risque faible à modéré sans prédicteur) et PMA (approbation préalable à la mise en marché, pour la classe III à risque le plus élevé). La grande majorité des dispositifs d'IA — environ 97 % — entrent par 510k); seulement quelques dizaines ont utilisé De Novo et une poignée d'AMP (Analyse de la liste de la FDA). Le point de repère De Novo était IDx-DR en 2018.
Le plan de contrôle du changement (PCCP), qui a été finalisé en décembre 2024, a été élaboré récemment.FDA). Un PCCP permet à un fabricant de pré-déterminer et d'autoriser un ensemble de modifications futures du modèle — et les propres mots de l'agence capturent la valeur: la FDA examine le PCCP "pour assurer la sécurité et l'efficacité continues de l'appareil sans nécessiter de soumissions de marketing supplémentaires pour mettre en œuvre chaque modification." En janvier 2025, la FDA est allée plus loin, publiant un projet complet d'orientation sur le cycle de vie total des produits du logiciel d'appareils compatibles avec l'IA. La posture des États-Unis, en bref: rapide, motivé par le prédicat, l'imagerie lourde, et maintenant organisé autour de la surveillance du cycle de vie.
Union européenne — deux régimes empilés sur un seul dispositif
L'UE est le marché le plus important de l'IA SaMD, parce que deux régimes réglementaires s'appliquent maintenant immédiatement. Premièrement, le Règlement sur les dispositifs médicaux. Sa règle de classification des logiciels — Règle 11 — est notoire. Lisez-le directement: "Le logiciel destiné à fournir des informations qui sont utilisées pour prendre des décisions à des fins diagnostiques ou thérapeutiques est classé dans la classe IIa", passant à classe III où une mauvaise décision pourrait causer la mort ou une détérioration irréversible, ou classe IIb pour une détérioration grave (MDR, annexe VIII, via EUR-Lex). Selon les anciennes directives, la plupart des logiciels autonomes auto-certifiés de classe I, sans aucune tierce partie. La règle 11 a poussé presque tous les diagnostics et thérapeutiques SaMD jusqu'à Classe IIa ou plus, qui force une Notified Body évaluation de la conformité, gestion de la qualité ISO 13485 et évaluation clinique. Notre propre scan du EUDAMED la base de données a trouvé des dispositifs AI-keyword concentrés dans la classe IIa — exactement l'empreinte de la règle 11.
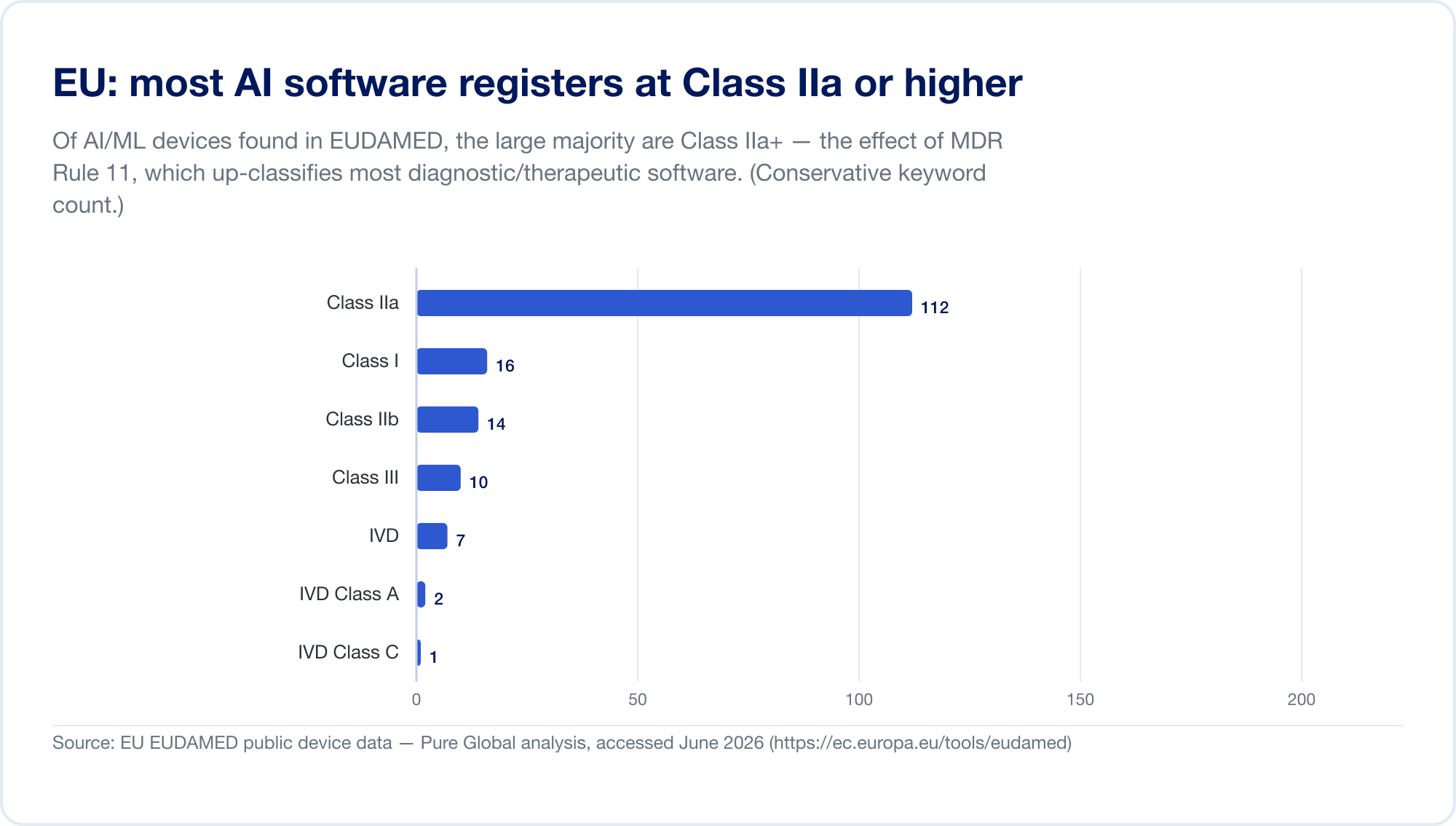
Puis, en couches sur le dessus, l'Acte d'IA de l'UE (Règlement 2024/1689), en vigueur depuis le 1er août 2024. Par l'article 6, paragraphe 1, un système d'IA est "à haut risque" lorsqu'il s'agit (ou est un élément de sécurité) d'un produit qui nécessite déjà une évaluation de la conformité par une tierce partie — qui saisit essentiellement toutes les catégories IIa+ Dispositifs médicaux de l'IA. La phase des obligations à haut risque s'étend jusqu'au 2 août 2026, la date d'introduction de l'IA dans les dispositifs MDR/IVDR étant fixée au 2 août 2027 (une proposition « Digital Omnibus » de novembre 2025 peut reporter cette date à 2028 — considérer les dates comme étant en mouvement) (intelligence artificielleact.eu). Les pénalités s'élèvent à 35 millions d'euros ou 7% du chiffre d'affaires mondial (Le présent règlement est obligatoire dans tous ses éléments et directement applicable dans tout État membre.). Pour clarifier ce chevauchement, le MDCG et le nouveau Conseil européen de l'IA ont publié une FAQ conjointe, MDCG 2025-6, en juin 2025 (Commission européenne).
La contrainte contraignante est la capacité. Le nombre d'organismes notifiés du MDR est passé d'environ 80–96 en vertu des directives à environ 50, avec seulement ~17–19 désignés en vertu du IVDR; la certification du MDR prend maintenant 13–18 mois en moyenne, soit environ le double de la norme antérieure au MDR (MedTech Europe). Pour AI SaMD, chaque appareil rivalise pour les rares Notified Body les créneaux horaires — et après 2027/28, il faudra aussi évaluer la conformité à la loi sur l'IA.
Royaume-Uni — divergent, pragmatique
Après le Brexit, le MHRA a tracé un parcours délibérément propice à l'innovation. Son programme Logiciel et IA en tant que programme de changement d'appareil médical (feuille de route publiée en octobre 2022) couvre onze paquets de travail, et l'agence s'est engagée à autoriser les PCCP dans les règles précommercialisation à venir (MHRA). Son bac à sable réglementaire AI Airlock, le premier du genre pour les dispositifs médicaux de l'IA, a réalisé un projet pilote de quatre projets en 2024 et une phase 2 de sept technologies jusqu'en 2026, avec un financement pluriannuel maintenant engagé (MHRA). Pratiquement, environ 90 % des appareils sur le marché britannique portent toujours la marque CE, que la Grande-Bretagne acceptera jusqu'au 2028-2030; le MHRA a consulté au début de 2026 sur la reconnaissance des marques CE indéfiniment (MHRA).
Canada — d'abord pour finaliser les règles spéciales de la LM
Santé Canada a été l'un des premiers organismes de réglementation à avoir reçu défini des directives précommercialisation spéciales pour les instruments médicaux à apprentissage automatique — finalisées pour la première fois en 2025 et publiées sous forme définitive révisée en avril 2026 — couvrant la classe II–IV, adoptant les termes clés du cadre de référence et introduisant officiellement le PCCP afin que les modifications autorisées ne déclenchent pas une nouvelle modification de licence (Santé Canada Le Canada a co-écrit les documents de base du trirégulateur – GMLP (2021), les principes PCCP (2023) et les principes de transparence (2024) – et a exigé la certification du PASMD depuis 2019.
Australie — réforme rapide, recalibrage pour AI
L'Australien TGA a réformé ses règles logicielles en février 2021, sculptant des applications de bien-être à faible risque tout en up-classifiant logiciel de diagnostic (appareils actifs pour la thérapie avec une fonction de diagnostic déplacé à la classe III). Sa consultation de 2024, Clarifier et renforcer la réglementation de l'IA, a attiré plus de 600 intervenants et produit 14 résultats clés actuellement en cours d'élaboration (TGA). La voie de dépendance de la TGA, qui accepte les « régulateurs étrangers comparables », est un accélérant majeur, dont il est question ci-dessous.
Japon — construit pour l'itération
Le Japon réglemente SaMD en tant que "dispositifs médicaux programmés" et a sans doute le mécanisme le plus adapté à la mise à jour de l'IA de tout grand marché: IDATEN, en vigueur depuis septembre 2020, est la version japonaise d'un protocole de gestion du changement post-approbation, permettant aux fabricants de changer d'accord préalable à l'IA fréquemment mise à jour (PMDA). Combiné avec le DASH pour SaMD l'initiative et la voie prioritaire SAKIGAKE, le Japon a construit une infrastructure pour les logiciels qui évolue — bien que l'adoption soit modeste: seulement 51 SaMD sur la liste du PMDA en septembre 2025.
Corée du Sud — le mouvement le plus rapide
La Corée est le leader. Au-delà de ses 153 autorisations d'IA en 2025, elle a mis en place un cadre juridique conçu spécifiquement: la Digital Medical Products Act (DMPA), en vigueur en janvier 2025, introduit un mécanisme de changement de style PCCP et un SGQ numérique aligné sur les éléments de travail de l'IMDRF (Emergo). La Corée a également publié la première ligne directrice du monde pour les dispositifs médicaux générateurs d'IA en janvier 2025 et a approuvé son premier dispositif en avril 2026, et elle préside le groupe de travail IMDRF AI/ML. Si vous voulez voir où va la réglementation mondiale de l'IA, regardez Séoul.
Singapour — le centre de confiance
Le HSA de Singapour est bien au-dessus de sa taille en étant le régime de dépendance le plus efficace en Asie. Son guide logiciel, GL-04 (Révision 4, décembre 2025), couvre explicitement les appareils à apprentissage automatique tout au long du cycle de vie et nécessite une notification de changement lorsque les performances, les intrants ou le niveau de vision humaine d'un modèle d'IA changent (HSA). Principalement, HSA reconnaît cinq organismes de référence (FDA des États-Unis, organismes notifiés de l'UE, Santé Canada, TGA, Japon MHLW) et a estimé qu'environ 98 % des applications peuvent utiliser une voie abrégée; les appareils ayant deux autorisations préalables peuvent s'enregistrer par une voie « immédiate » en aussi peu qu'une heure (Singapour).
Chine — grande, distincte et exigeante
La NMPA chinoise traite sérieusement les logiciels d'aide à la décision en matière d'IA: ses principes de révision 2021 CMDE et son catalogue de classification 2021-2022 placent le logiciel qui délivre un diagnostic ou stimule le traitement en Class III, le niveau de risque le plus élevé. La Chine a approuvé son premier dispositif AI de classe III en 2020 et avait atteint environ 154 dispositifs médicaux AI au milieu de 2025 (JMIR Informatique médicale). Dans un paquet de réformes d'octobre 2025, la NMPA s'est engagée à "simplifier les exigences d'enregistrement de changement pour les dispositifs médicaux à moteur AI où l'algorithme de base reste inchangé mais où la performance de l'algorithme est optimisée" — une concession réelle mais étroite par rapport au PCCP américain (NMPA). La Chine a besoin d'un agent dans le pays, de tests de type locaux et, pour de nombreux appareils de classe III, de données cliniques locales, ce qui en fait l'un des marchés les plus exigeants du présent rapport.
Inde, Taïwan et le reste de l'Asie-Pacifique
En octobre 2025, le CDSCO a publié un projet de guide sur les logiciels d'appareils médicaux, introduisant un « Protocole de changement d'algorithme » pour les mises à jour de l'IA, mais il demeure un projet sans règle d'IA finalisée (CDSCO Taiwan's TFDA, en revanche, a l'une des suites de guidage AI les plus profondes n'importe où — directives techniques CADe/CADx dédiées et directives de rédaction PCCP — et 166 dispositifs AI/ML autorisés de 2020 à 2024. Dans l'ensemble de l'ANASE, la tendance est la dépendance-plus-localisation: Malaisie, Thaïlande, Vietnam, Philippines et Indonésie traitent principalement SaMD en tant que dispositif général et s'appuyer sur les approbations de pays de référence, avec la voie rapide du Vietnam exceptionnellement large (il accepte même les approbations NMPA et MFDS).
Amérique latine, Moyen-Orient et Afrique
Le Brésil ANVISA a importé la logique de gros de l'UE: sa règle 11 sous RDC 751/2022 reflète l'UE, plaçant le logiciel d'aide à la décision dans la classe II-IV, et RDC 657/2022 a été le premier SaMD- résolution spécifique. Un fabricant étranger ne peut pas détenir d'enregistrement brésilien - un titulaire local d'enregistrement est obligatoire et non disponible (Artixio La COFEPRIS du Mexique a transformé son régime de dépendance en 2025 en une seule voie abrégée reconnaissant tous les membres de l'IMDRF et du MDSAP, ciblant 30 jours ouvrables. La SFDA de l'Arabie saoudite a publié MDS-G010 (novembre 2022) — parmi les premières directives médicales dédiées à l'IA/ML n'importe où, et citée par certains observateurs comme étant la première enforceable (autres la classent comme non contraignante) — ce qui indique uniquement que "le fabricant devrait valider localement les dispositifs médicaux basés sur l'IA/ML qui ont été développés et approuvés dans d'autres juridictions" (SFDA) — un rappel que "l'approbation du pays de référence" n'est pas toujours suffisante. L'approbation centralisée des dispositifs EAU dans le cadre d'un nouvel Emirates Drug Establishment en janvier 2025. La SAHPRA de l'Afrique du Sud a publié sa première communication AI en septembre 2025 mais n'a pas encore commencé à enregistrer les dispositifs, et la continentale Agence Africaine des Médicaments — 31 des 55 États ratifiés — ne couvre pas encore les dispositifs ou l'IA.
C'est le cœur du problème: quinze juridictions, quinze réponses. Le même logiciel est la classe II aux États-Unis, la classe IIa+ et « à haut risque » dans l'UE, la classe III en Chine, la classe 2–3 en Corée et la classe II–IV au Brésil, chacun ayant ses propres exigences en matière de preuve, de langue, de propriétaire local et de contrôle du changement.
Ce que ça coûte et combien de temps ça prend
Les différences de classification ci-dessus se traduisent directement en argent et en mois. Les chiffres principaux ci-dessous sont les frais du gouvernement et les délais réalistes pour un appareil à risque élevé; ils excluent les coûts substantiels des tests, des preuves cliniques, de la traduction et de la représentation dans le pays qui souvent nainent les frais officiels.
La taxe officielle est le petit nombre
À lui seul, les frais du gouvernement vont de moins de 1 000 à environ 44 000 dollars pour une inscription à haut risque:
- États-Unis — MDUFA FY2026 droits (vérifiés sur FDA.gov): 510k) 26 067$ (petite entreprise 6 517); De Novo 173 782$; APM 579 272$; plus un droit d'établissement annuel de 11 423$ (FDA).
- Chine — Les frais d'enregistrement de la NMPA sont d'environ 210 900 (~30 000 $) pour la catégorie II et 308 800 (~44 000 $) pour la catégorie III — les frais officiels les plus élevés du présent rapport.
- Brésil — ANVISA classe III/IV enregistrement de ~BRL 21 000 plus une taxe de certification B-GMP de BRL 72,804 pour les fabricants internationaux.
- Canada — Catégorie III CAD 14 163$, Catégorie IV CAD 30 713$ (2026).
- Arabie saoudite — Honoraires de la SFDA de 15 000 à 23 000 SAR par classe.
- Inde — Licence d'importation MD-15 de 3 000 $ par site + 1 500 $ par produit pour la classe C/D.
- Singapour, Australie, Corée, Japon - les honoraires officiels sont relativement modestes (souvent inférieurs à 13 000 dollars), mais le fardeau de la preuve et de l'examen varie considérablement.
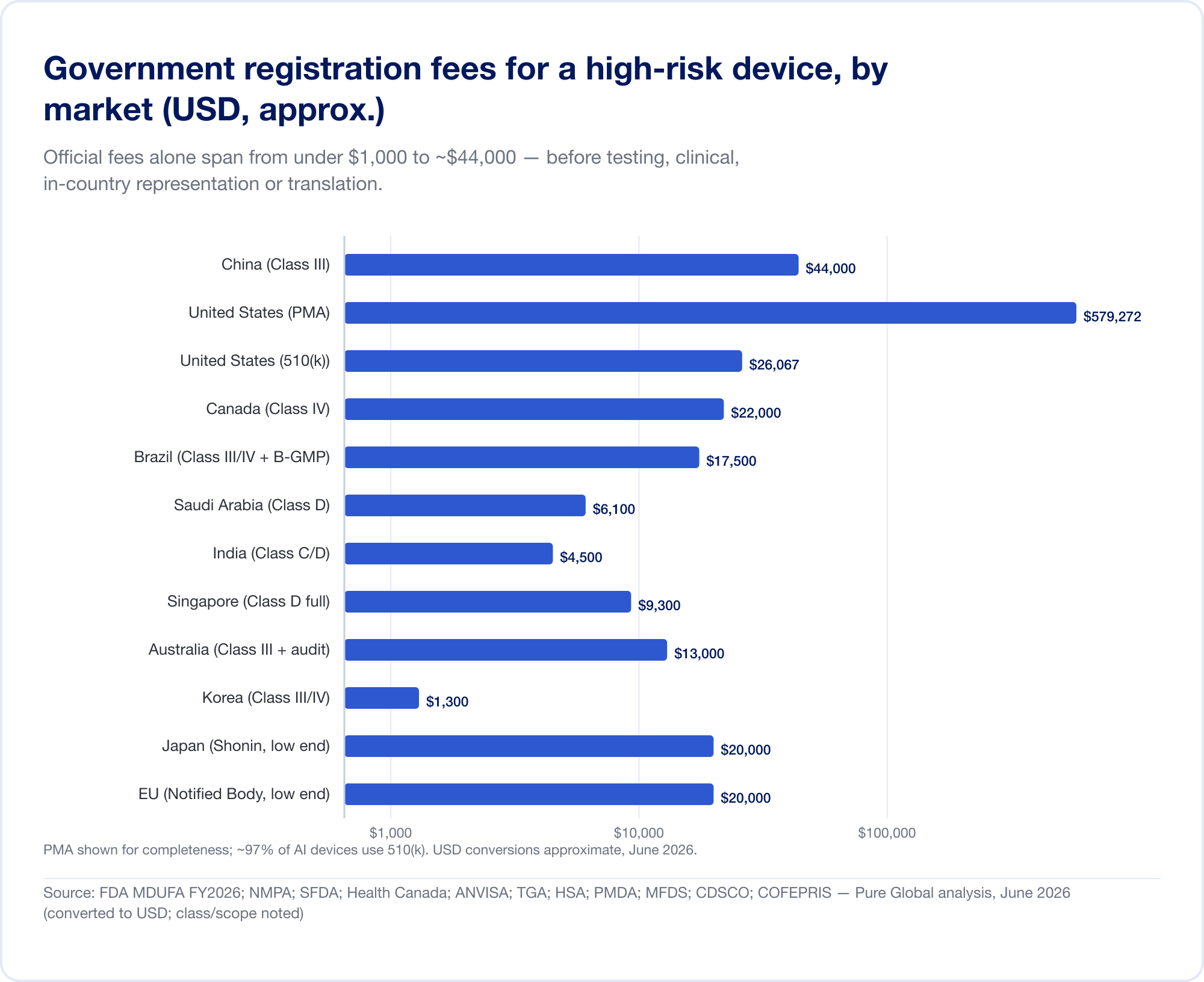
Le temps est le chiffre cher
Le coût réel est le calendrier. Les délais réalistes et à risque élevé vont d'un mois environ (Canada, catégorie III) à 24 mois dans l'UE et quatre à cinq ans en Chine lorsqu'un essai clinique local est nécessaire:
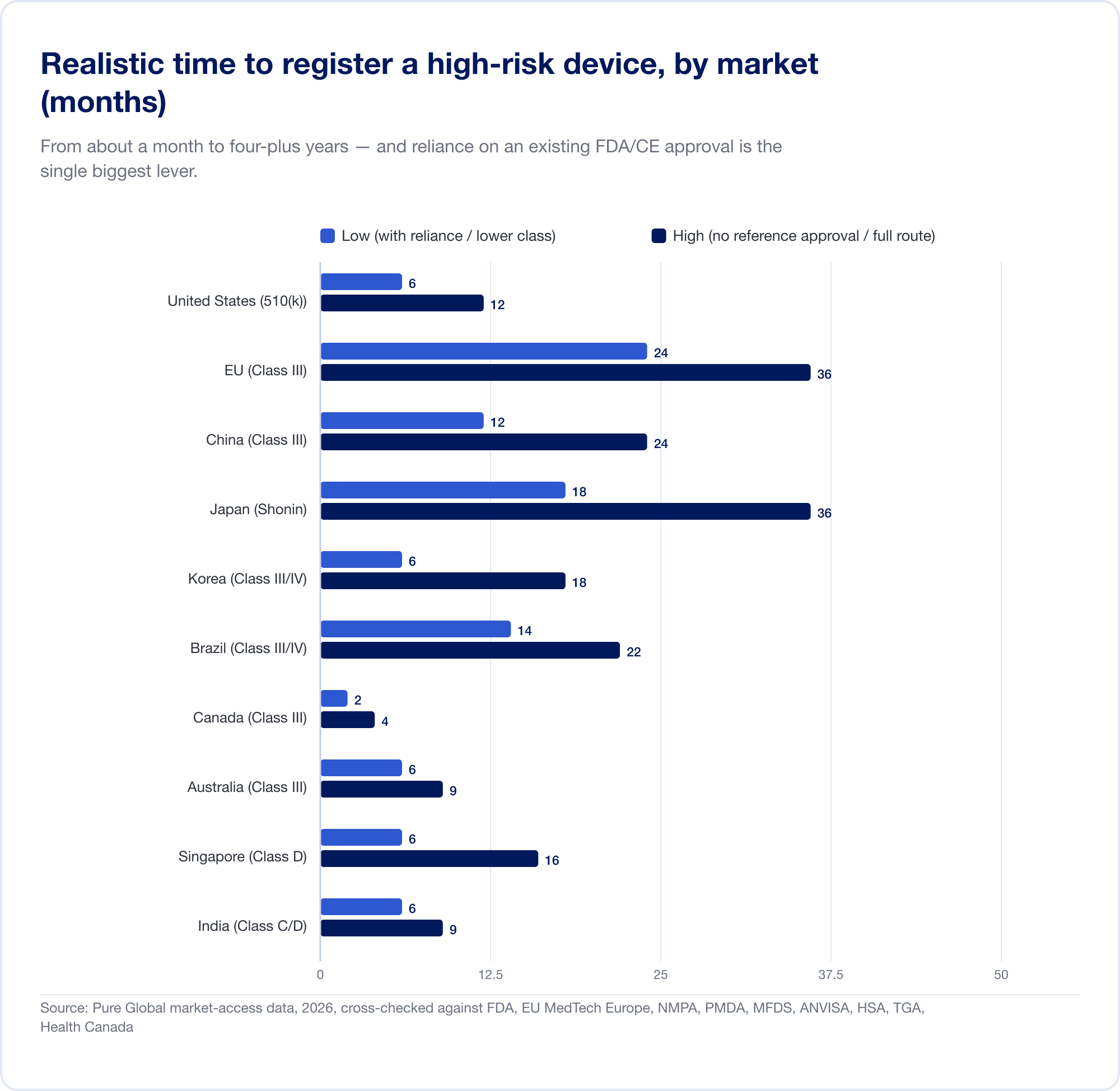
L'UE illustre comment la classification devient un coût. Parce que la règle 11 convertit ce qui était une autodéclaration de classe I quasi libre en une déclaration de classe IIa+Notified Body l'engagement, le projet CE SaMD Il s'agit généralement de six chiffres et 13–18 mois avant la délivrance d'un certificat. Pour une startup avec une piste mesurée en quartiers, ce n'est pas un élément de ligne — c'est une menace stratégique.
Le levier: la dépendance et la torsion spécifique à l'IA
En dépit de ces délais, c'est l'outil le plus puissant dans l'accès aux marchés mondiaux: dépendance. La plupart des marchés en dehors des États-Unis, de l'UE et de la Chine s'appuieront sur une approbation que vous avez déjà. Nos propres données de marché montrent clairement l'effet — un risque élevé SaMD qui prend ~8 mois pour s'inscrire au Brésil sur la route standard peut descendre vers 6 semaines quand une FDA ou une autre approbation de référence est mise à profit par la voie d'analyse optimisée d'ANVISA.
Pour AI SaMD il y a une torsion spécifique et récurrente que les tableaux de coûts ne montrent pas. Lorsque l'algorithme est mis à jour — comme l'IA le fait constamment — la question est de savoir si cette mise à jour nécessite une soumission nouvelle. Le PCCP des États-Unis peut laisser les mises à jour pré-spécifiées expédier sans nouvelle soumission, en économisant les frais 26 067 $ plus l'examen de 90–175 jours sur chaque évité 510(k), et beaucoup plus pour éviter les suppléments APM. Environ 10% des dispositifs d'IA que la FDA a nettoyés en 2025 ont déjà intégré un PCCP. Mais cette épargne n'est qu'aux États - Unis — et c'est la charnière de tout le rapport.
Combien coûte l'enregistrement de l'IA en tant qu'instrument médical dans différents pays?
Les frais gouvernementaux ci-dessus ne sont que la moitié de la facture. L'autre moitié est le travail de spécialiste: construire le dossier, tenir l'enregistrement local, poser toutes les questions de l'autorité, et déposer chaque renouvellement et changement d'algorithme. C'est là que l'industrie est la plus opaque: la plupart des cabinets de consultants réglementaires facturent à l'heure ou citent chaque présentation séparément, de sorte que le coût réel multi-marchés n'apparaît qu'après l'arrivée des ordres de modification.
Pure Global est la première entreprise d'accès au marché des dispositifs médicaux à publier un tarif annuel unique et forfaitaire par enregistrement. À partir de 2 000 $US par appareil, par marché, par année, un droit consolide les services facturés normalement à l'heure — représentation dans le pays, présentation (sur approbation de référence), renouvellements, modifications et toute correspondance entre les autorités sanitaires. Pas de feuilles de temps, pas de surprises par mail.
Voici exactement ce que ça coûte d'avoir Pure Global agir en tant que représentant local et détenir un enregistrement de dispositifs médicaux AI, marché par marché. (AI)SaMD En général, les terres appartiennent à la catégorie à risque élevé, de sorte que le chiffre supérieur s'applique aux marchés à paliers.)
Pure Global Représentation dans le pays — frais annuels forfaitaires par appareil d'IA
- Un numéro transparent par marché, par an — tout pour cette inscription inclus.*
| Marché | Rôle local Pure Global fournit | Taxe annuelle forfaitaire (USD) |
|---|---|---|
| États-Unis | FDA Agent américain | $1,000 |
| Union européenne | EUAuthorized Representative | $2,000 |
| Royaume-Uni | Personne responsable au Royaume-Uni (UKRP) | $2,000 |
| Australie | Sponsor TGA | $2,000 |
| Singapour | Titulaire | 2 000 $ · 3 000 $ (classe C/D) |
| Malaisie | Authorized Representative | 2 000 $ · 3 000 $ (classe C/D) |
| Thaïlande | Authorized Representative | 2 000 $ · 3 000 $ (classe 3/4) |
| Indonésie | Authorized Representative | $2,000 |
| Vietnam | Titulaire de l'autorisation de mise sur le marché | $2,000 |
| Hong Kong | Responsable local | 2 000 $ · 3 000 $ (classe III/IV) |
| Macao | Titulaire de la licence & enregistrement | 2 000 $ · 3 000 $ (classe III) |
| Brésil | Titulaire de l'enregistrement au Brésil (BRH) | 2 000 $ · 3 000 $ (classe III/IV) |
| Mexique | Titulaire de l'enregistrement au Mexique | 2 000 $ · 3 000 $ (classe II/III) |
| Colombie | Représentant d ' INVIMA | 2 000 $ · 3 000 $ (classe IIb/III) |
- Frais annuels de représentation d'une AI dans le pays SaMD; comprend la présentation sur approbation de référence, les renouvellements, les modifications et la correspondance d'autorité. Source:Pure Global Liste de prix principale, 2026 (par inscription; des remises multi-inscriptions et 3 ans de contrat s'appliquent).*
Une compilation américaine 510(k) fonctionne de 15 000 $ à 20 000 $, un projet de documentation technique ou d'URCE de l'UE est répertorié par classe (8 000 $ à 30 000 $), une compilation canadienne d'enregistrement est de 3 000 $ à 25 000 $ par classe, et une détermination de la voie réglementaire est plate 5 000 $. Chaque chiffre est cité à l'avance, jamais à l'heure.
Un exemple concret — un algorithme d'imagerie de l'IA, quatre marchés. Prenez un seul outil de radiologie AI déjà autorisé par la FDA (classe II) et CE-marqué (classe IIb) que vous voulez maintenir en vie à travers les États-Unis, l'Union européenne, le Brésil et Singapour pendant un an.Pure Global La représentation dans le pays totalise 1 000 $ (É.-U.) +2 000 $ (UE) + 3 000 $ (Brésil) + 3 000 $ (Singapour) = 9 000 $ pour l'année – appartement, avec chaque renouvellement, modification et échange d'autorité inclus; vous ajoutez un travail de soumission unique seulement lorsqu'un marché nécessite effectivement un nouveau dossier. Cette prévisibilité est le point: lorsque les règles diffèrent dans chaque marché et que votre modèle change constamment, la dernière chose dont un fabricant a besoin est un projet de loi réglementaire qui le fait aussi.
Le paradoxe de la convergence
Voici la bonne nouvelle apparente. Sous le patchwork à quinze juridictions fonctionne une puissante machine de harmonisation. L'IMDRF aligne les définitions et les principes de bonnes pratiques d'apprentissage automatique. Le Medical Device Single Audit Program (MDSAP) permet à un seul système de vérification de la qualité de satisfaire cinq organismes de réglementation à la fois — les États-Unis, le Canada, le Brésil, le Japon et l'Australie (FDA). Et les voies de dépendance se propagent rapidement: Singapour accepte cinq agences de référence et les voies ~98 % des demandes par un examen abrégé; le Brésil, le Mexique, l'Australie, la Malaisie, le Vietnam et le Golfe reconnaissent tous les approbations étrangères dans une certaine mesure. En février 2026, l'IMDRF a même publié un manuel de référence pour codifier la pratique (IMDRF N89).
Quels marchés acceptent les autorisations étrangères (itinéraires de dépendance)
Une autorisation de la FDA ou une marque CE est une clé principale pour des dizaines de marchés, chacun avec sa propre serrure.
| Marché | Organismes/programmes de référence reconnus | Effet |
|---|---|---|
| Singapour (HSA) | États-Unis FDA, UE NB, Santé Canada, TGA, Japon MHLW | Abréviation/expédiée/immédiate; environ 98 % admissible |
| Brésil (ANVISA) | TGA, Santé Canada, FDA américaine, Japon MHLW (classe III/IV) | "Analyse optimisée" ~20-30% plus rapidement |
| Mexique (COFEPRIS) | Tous les membres de l'IMDRF + participants du PAIM | Voie abrégée, 30 jours ouvrables |
| Australie (TGA) | FDA des États-Unis, Santé Canada, MHLW/PMDA, EU NB, MDSAP | Évaluation de la conformité abrégée |
| Malaisie (MDA) | États-Unis FDA, Santé Canada, TGA, UE CE, PMDA, HSA, Thaïlande FDA | Voie de vérification (abréviation) + PASMD |
| Vietnam (MOH) | FDA des États-Unis, UE, PMDA, TGA, Santé Canada, MFDS, NMPA | Piste rapide SRA exceptionnellement large |
| Arabie saoudite (SFDA) | FDA/CE appuyant uniquement | Un examen complet du dossier technique reste nécessaire |
| EAU (EDE) | CE, FDA américaine | Enregistrement fondé sur la fiabilité |
| PAIMD (une vérification) | États-Unis, Canada, Brésil, Japon, Australie | Vérification unique du SGQ acceptée par les cinq |
Source: HSA, ANVISA, COFEPRIS, TGA, MDA Malaisie, FDA thaïlandaise, Vietnam MOH, SFDA, EDE —Pure Global analyse, juin 2026.
Pour un dispositif statique, il s'agit d'une transformation: une forte approbation – généralement la FDA ou le CE – devient une clé maîtresse qui ouvre des dizaines de marchés à une vitesse et à un coût réduits. C'est précisément le levier qu'un programme d'accès au marché bien géré est conçu pour l'exploiter.
Et voici le paradoxe. Pour l'AI adaptée, la clé maîtresse cesse de fonctionner exactement au point le plus important — le contrôle du changement. La convergence est sur l'instrument*; la divergence est sur l'AI*. Considérez le même modèle d'apprentissage automatique qui cherche à mettre à jour son algorithme:
- Dans le U.S., un PCCP préautorisé le laisse expédier la mise à jour sans nouvelle soumission.
- Dans le EU, un changement de logiciel "important" déclenche toujours Notified Body révision — et, en couches sur le dessus, évaluation distincte de la conformité de la loi sur l'IA.
- En Chine, la mise à jour n'est tolérée que si « l'algorithme de base reste inchangé »; un véritable recyclage signifie l'enregistrement complet du changement.
- Dans Corée, la DMPA autorise les plans de changement préapprouvés, mais seulement dans les paramètres préapprouvés.
Comme l'indique une analyse, "un PCCP autorisé par la FDA ne satisfait pas aux obligations de la loi sur l'IA de l'UE, et vice versa" (Berkley Sciences de la vie). Aucune vérification du PASMD et aucune voie de dépendance ne résout cette situation. Un développeur d'IA qui gagne des autorisations sur dix marchés n'a pas acheté la paix; ils ont acheté dix obligations différentes de contrôle du changement, chacune déclenchée chaque fois que le modèle s'améliore. Pour une technologie dont la totalité de la proposition de valeur est qu'elle continue à s'améliorer, c'est-à-dire une taxe structurelle qui compense le succès — et elle tombe plus durement sur les petites équipes qui construisent les meilleurs modèles.
La prochaine frontière: modèles génériques d'IA et de fondation
Si l'IA adaptative a tendu le système, l'IA générative menace de l'écraser. Tout ce qui précède suppose un modèle formé pour une seule utilisation prévue bien définie: détecter un saignement, mesurer une fraction d'éjection, signaler un nodule. Les grands modèles linguistiques et les modèles de base multimodaux rompent cette hypothèse de trois façons à la fois: ils sont à usage général (un modèle, de nombreuses utilisations possibles), non déterministes (la même prompte peut donner des réponses différentes), et sujets à hallucination (confidentiel, couramment, faux). Aucune de ces propriétés ne s'inscrit confortablement dans un cadre construit autour d'une utilisation prévue fixe et d'une version de référence « verrouillée ».
Les régulateurs le savent. Le OMS a publié en janvier 2024 la première ligne directrice mondiale consacrée aux grands modèles multimodal, mettant en garde plus particulièrement contre les sorties fabriquées, les biais d'automatisation et la difficulté de valider les systèmes formés sur les données à l'échelle d'Internet (WHO). Le Comité consultatif sur la santé numérique de la FDA a consacré sa réunion inaugurale, en novembre 2024, aux défis du cycle de vie total des dispositifs générateurs d'IA (FDA). Et La Corée du Sud, caractéristiquement la première, a publié la première ligne directrice du monde pour les dispositifs médicaux generative-AI en janvier 2025 et a autorisé son premier dispositif en avril 2026.
Mais l'orientation n'est pas la même qu'une voie de dégagement. Au milieu des années 2026, les itinéraires établis — 510(k), De Novo, marquage CE — supposent toujours un dispositif que vous pouvez épingler, tester contre une norme fixe et geler. Une LLM clinique d'usage général ne satisfait aucune de ces conditions préalables proprement, c'est pourquoi la première vague de « l'IA générative dans les soins de santé » a atteint le marché principalement en tant qu'outils administratifs (scribes d'ambients, aides à la documentation) qui s'écartent de la définition de l'appareil, plutôt qu'en tant que dispositifs de diagnostic nettoyés. La frontière réglementaire pour l'IA clinique autonome et générative est, vraiment, encore en train d'être tracée — et les marchés qui la dessinent d'abord (Corée aujourd'hui, d'autres à suivre) façonneront comment le reste du monde les copie. Pour les développeurs, la leçon pratique est de regarder où les lignes sont fixées et de concevoir la stratégie réglementaire, pas seulement le produit, pour une cible mobile.
Le playbook d'accès au marché
Si le problème est la fragmentation, la réponse est un système. À travers les modèles ci-dessus, un playbook répétable émerge pour obtenir un appareil médical d'IA dans le monde — et le garder là.
Classez avant de construire les preuves. Le même produit peut être de classe II ou III selon le marché et l'allégation. Cartez les règles de classification des marchés cibles premier, car elles dictent les preuves cliniques et techniques dont vous aurez besoin. Le séquençage de la réclamation et des éléments de preuve sur le marché cible le plus strict évite la reconstitution du dossier plus tard.
Gagnez une forte approbation d'ancrage, puis exploitez la confiance délibérément. Une autorisation de la FDA ou une marque CE vaut bien plus d'un marché; c'est le titre qui déverrouille les routes abrégées à Singapour, au Brésil, au Mexique, en Australie, dans le Golfe et au-delà. L'art est de savoir qui ancre chaque marché cible reconnaît, et d'orienter le dossier en conséquence. L'Arabie saoudite, qui exige une validation locale même de l'IA approuvée par l'étranger, rappelle que la dépendance est une carte et non une couverture.
Défendre la représentation dans le pays où elle est obligatoire, ce qui est la plupart des lieux. Un fabricant étranger ne peut pas détenir sa propre immatriculation au Brésil (BRH), l'UE (Authorized Representative), la Chine (agent juridique), le Japon (MAH/DMAH), l'Arabie saoudite, les Émirats arabes unis, l'Inde, et bien d'autres encore. Chacun d'eux exige qu'une personne morale locale tienne l'enregistrement et fasse front à l'autorité sanitaire. Il n'est pas pratique de compter trente entités de ce type; l'impartition de ces entités à un seul partenaire est la façon dont l'échelle devient réalisable.
Traiter le contrôle du changement comme une filière de travail multimarchés de première classe. C'est la discipline spécifique à l'IA. Construisez un PCCP américain, mais aussi map comment chaque marché gère la mise à jour même — et concevoir la cadence de libération de l'algorithme autour du régime le plus restrictif qui compte pour votre entreprise. Le plan du cycle de vie fait maintenant partie du plan d'accès au marché.
Exécutez-le comme une seule opération connectée, pas trente dépôts déconnectés. Le coût n'est pas un seul enregistrement; il s'agit de la coordination — traductions, détenteurs locaux, calendriers de renouvellement, notifications de changement et vigilance post-commercialisation à travers des dizaines de régimes qui dérivent indépendamment.
Une séquence typique en pratique. Pour un développeur d'imagerie-AI, le déploiement fonctionne souvent: sécurisez la marque FDA 510(k) ou CE en tant qu'ancrage; en parallèle, déposez sur votre marché d'origine et un marché à forte dépendance (Singapour ou Australie) pour obtenir des revenus anticipés; utilisez ces autorisations pour ouvrir les routes abrégées au Brésil, au Mexique et dans le Golfe; puis prenez sur les marchés de haute valeur - Chine et Japon - où les tests locaux ou les données cliniques sont inévitables et les délais sont plus longs. Tout au long, un seul plan de contrôle du changement est maintenu au niveau central et cartographié selon les règles de mise à jour de chaque marché, de sorte qu'une amélioration de modèle est déposée partout où elle doit être, et nulle part où elle n'est pas nécessaire. L'ordre n'est pas arbitraire; il suit délibérément les flux de trésorerie, la réutilisation des preuves et les marchés à ressources limitées.
C'est le travail Pure Global est conçu pour: la représentation dans le pays et l'exécution de la réglementation assistée par AI sur les marchés 30+, livrée sur un tarif annuel fixe plutôt que sur le modèle horaire ouvert auquel l'industrie est par défaut. Les données du présent rapport — tirées de la FDA,EUDAMED, NMPA, PMDA, MFDS, ANVISA et des dizaines de registres nationaux, à côté de notre propre ensemble de données sur les coûts et les délais de commercialisation, est la même intelligence que nous utilisons pour séquencer le déploiement global d'un client. Le point de cartographier le labyrinthe est de pouvoir le parcourir rapidement.
Conclusion: quatre choses à emporter
L'AI est maintenant une catégorie d'appareils grand public, et les données le prouvent. D'environ 1 sur 700 autorisations FDA en 2019 à 1 sur 28 en 2025, avec 1 500 dispositifs AI plus autorisés aux États-Unis seuls et programmes nationaux en Corée, en Chine, à Taïwan et au Japon à l'échelle rapide, AI SaMD est passé de la nouveauté à la norme.
Chaque organisme de réglementation le classe à la hausse, et la plupart d'entre eux convergent sur la surveillance du cycle de vie. Règle 11 dans l'UE et au Brésil, classe III en Chine, risque élevé en vertu de l'AI Act de l'UE — l'IA diagnostique et thérapeutique est traitée comme grave, et la réponse partagée est le contrôle du cycle de vie total des produits, les GMLP et les plans de changement prédéterminés.
La confiance effectue un voyage d'approbation — sauf pour la partie AI. Le mécanisme d'harmonisation (IMDRF, MDSAP, itinéraires de dépendance) permet véritablement une forte approbation d'ancrage débloque des dizaines de marchés. Mais le contrôle adaptatif du changement diverge fortement, de sorte qu'une seule approbation ne reste pas valide partout au fur et à mesure que le modèle évolue. Cette lacune est le défi opérationnel déterminant du terrain.
L'avantage concurrentiel est la machine d'enregistrement, et non l'enregistrement. Lorsque les règles diffèrent sur chaque marché et changent chaque année, l'avantage durable appartient à ceux qui peuvent s'enregistrer rapidement, partout, et garder chaque approbation vivante à travers chaque mise à jour modèle — comme un système connecté.
Parle-nous.
Si vous construisez ou développez un appareil médical d'IA et que vous pèsez sur quels marchés entrer, dans quel ordre et comment maintenir chaque approbation valide à mesure que votre modèle s'améliore, c'est exactement le problème que nous résolvons.Parler à Pure Global un plan d'accès au marché basé sur les données ci-dessus — ou Guides d'enregistrement marché par marché pour aller plus loin dans n'importe quel pays.
Sources
Gouvernement et organismes d'harmonisation cités ci-dessus; références clés regroupées ci-dessous. Tous les chiffres sont datés; les chiffres de la taille du marché et des prévisions sont des estimations de tiers dont la portée varie et devrait être interprétée comme des fourchettes.
Définitions, cadres et principes directeurs
- IMDRF — SaMD: Définitions clés (N10, 2013); * catégorisation des risques* (N12, 2014); dispositifs médicaux compatibles avec les LM: termes clés (N67, 2022); principes directeurs des LMG (N88, 2025); livre de lecture de la fiabilité (N89, 2026). imdrf.org
- US FDA — Logiciel comme instrument médical (SaMD Cadre réglementaire proposé pour les modifications à l'IA/ML SaMD (2019); Plan d'action AI/ML (2021). Conseil
- OMS — Éthique et gouvernance de l'IA pour la santé (2021); * Considérations réglementaires relatives à l'IA pour la santé* (2023); Directives sur les MLM (2024); Bonnes pratiques de fiabilité, TRS 1033 Annexe 10 (2021). qui.int
États-Unis
- FDA — * Liste des instruments médicaux à base d'intelligence artificielle* (mise à jour du Q1-2026, 1 524 appareils; ~76 % de radiologie); Guide final du PCCP (déc. 2024); Comité consultatif sur la santé numérique sur l'IA générative (nov. 2024); MDUFA FY2026 frais. fda.gov · Le fil d'imagerie (analyse de partage de radiologie, 2026); Innolitics & IntuitionLabs (trackers de compensation, 2025–26).
Union européenne et Royaume-Uni
- EUR-Lex — Règlement (UE) 2017/745 (MDR), annexe VIII, règle 11; règlement (UE) 2024/1689 (AI Act), articles 6, 99 et 113.MedTech Europe & Équipe-NB (Notified Body capacité). MHRA — Programme de changement de logiciel et d'IA; AI Airlock; guide de reconnaissance CE. Conseil
Canada, Australie, Japon, Corée
- Santé Canada — Directives préalables à la mise en marché pour les instruments médicaux compatibles avec les LM (avril 2026). canada.ca
- TGA — réformes logicielles (2021); résultats de la consultation sur l'IA (2024–26). Tga.gov.au
- PMDA/MHLW —SaMD des conseils; DASH pour SaMD IDATEN. pmda.go.jp
- MFDS — 2025 Rapport d'approbation (153 dispositifs d'IA); DMPA; ligne directrice generative-AI. mfds.go.kr; bioin.or.kr
Asie-Pacifique, Latam, MEA
- HSA Singapore — GL-04-R4; agences de référence et de confiance. Autres
- NMPA Chine — Classification AI et réforme oct 2025. nmpa.gov.cn · JMIR Medical Informatics (154 AIMD, 2026).
- CDSCO Inde — Projet de guide logiciel MD (octobre 2025). TFDA Taiwan — Guide CADe/CADx; J. Formos. Med. Assoc. (166 licences).
- ANVISA Brésil — RDC 751/2022 (règle 11), RDC 657/2022, IN 290/2024. gov.br/anvisa
- COFEPRIS Mexique — Voie abrégée (2025). SFDA Arabie saoudite — MDS-G010 (2022). EAU EDE — Décret-loi fédéral 38/2024. SAHPRA Afrique du Sud — Communication AI (2025).
Marché, entreprises, brevets, sécurité
- Taille du marché: Grand View Research; MarketsandMarkets; The Business Research Company; Mordor Intelligence; Fortune Business Insights (2025–26).
- Financement et entreprises: Rock Health (2025 financement de la santé numérique); CB Insights; société FY2025 dépôts (Tempus AI, iRhythm, HeartFlow, Butterfly Network, Lunit, VUNO); Aidoc (série E).
- Brevets: Tendances technologiques de l'OMPI (2019) et paysage génétique des brevets de l'IA (2024); CNIPA.
- Sécurité: NEJM (précisation de l'oxyde d'impulsion, 2020); JAMA Internal Medicine (Epic Sepsis Model, 2021); JAMA Health Forum & JAMA Network Open (Résumés de l'IA, 2025); guide de la FDA sur l'oxyde d'impulsion.
- Commerce: OCDE-FMI-OMC Mesure du commerce numérique (2021); OMC; CNUCED.
- Pure Global données exclusives sur les coûts d'accès au marché et les délais (2026); open FDA,EUDAMED Analyse de la base de données MFDS —Pure Global, juin 2026.



Parlons,
N'importe où vous êtes.
Que vous cherchiez plus d'information ou que vous soyez prêt à travailler en partenariat avec nous, nous sommes là pour vous guider à chaque étape du processus réglementaire.
Contactez-nous