
AI 作为医疗器械:全球监管、注册和市场准入地图
人工智能是历史上增长最快的一类医疗器械,也是监管差异最大的一类医疗器械。美国 FDA 现在列出了 1,524 个支持 AI 的设备;韩国一年授权153个。然而,同一个软件在美国是 II 级,EU 是 IIa+ 级和“高风险”,在中国是 III 级——每个软件都有自己的证据、本地持有者和变更控制规则。该报告描绘了每个主要监管机构如何将 AI 作为医疗器械进行分类、批准和监管,包括 30 多个市场的注册成本、时间表和依赖路线,以及无需每次重建档案即可实现这些目标的手册。
一份基于证据的现场指南,介绍世界监管机构如何对医疗保健领域的人工智能进行分类、批准和监管,以及如何在不每次都重建档案的情况下进入 30 多个市场。
长话短说
人工智能已成为历史上增长最快的医疗器械类别。美国食品和药物管理局公开的 AI 设备列表到 2026 年中期已达到 1,524 项授权,其中约四分之三属于放射学(FDA)。根据我们对 FDA 510(k) 数据库的分析,AI 在所有许可中所占的份额从 2019 年的约 1/700 上升到 2025 年的约 1/28。仅韩国一国就在 2025 年授权了 153 个 AI 设备(MFDS)。
但在一个机构中运行的同一个软件可能会在下一个机构中停滞不前。以下是这份报告的论点,主要有五点:
- AI 是一种设备,其中大部分是“软件作为医疗器械”(SaMD)。 当软件进行诊断、分类或推荐治疗时,它就像手术刀或扫描仪一样受到监管——在数十个司法管辖区,每个司法管辖区都有自己的规则。
- 全世界都对它进行了升级。 EU的MDR规则11、巴西的规则11、中国诊断AI的III级默认——几乎所有地方的诊断和治疗软件都属于较高风险类别,需要第三方审查。
- 自适应AI打破了一次性批准模型。 这种模型在发布后不断学习变化,因此监管机构发明了新的机制——美国的预定变更控制计划、日本的 IDATEN、韩国的 DMPA——这些机制彼此并不匹配。
- 信赖趋于一致,AI 则趋于分歧。 一张由信任和认可途径组成的网络应该可以让一次批准打开许多市场。它适用于静态设备。而对于自适应 AI,“FDA 授权的 PCCP 不满足 EU AI 法案义务,反之亦然”(Berkley Lifesciences)。
- 因此,获胜者将多市场注册产业化。 竞争优势不再是单一的清关;它是一台可操作的机器,可以将一项批准变成三十项批准,并随着算法的发展保持每一项批准都有效。
Pure Global 缩小了差距——国内代表和 AI 协助 30 多个市场的监管执行,并收取固定年费。本报告的其余部分是地图。
“AI 作为医疗器械”实际上是什么
从最重要的词开始: 装置。如果一款软件“旨在用于疾病的诊断、治疗或预防”,那么它就是地球上几乎每个法律体系中的医疗器械——无论它是在 MRI 扫描仪内的芯片上运行,还是作为放射科医生浏览器中的应用程序运行。 AI 在大多数国家没有自己的法规;它继承了医疗器械法的整个体系。
锚点的定义来自 国际医疗器械监管机构论坛 (IMDRF),主要机构协调其词汇的机构。在 2013 年的基础文件中,IMDRF 将软件作为医疗器械 (SaMD) 定义为“旨在用于一个或多个医疗目的的软件,无需成为硬件医疗器械的一部分即可执行这些目的” (IMDRF N10)。 FDA 逐字采用了该语言,并绘制了对下游所有事物都很重要的三向区别(FDA):
- SaMD — 软件 是 医疗器械(胸部 X 光分类算法、糖尿病视网膜病变探测器)。这是大多数临床AI 居住的地方。
- SiMD — 医疗器械中的软件,硬件的组成部分(运行输液泵的固件)。
- 软件用于 制造或维护 一个设备,再次受到不同的监管。
特别是对于 AI,IMDRF 的 2022 年关键术语文档将支持机器学习的医疗器械定义为“部分或全部使用机器学习来实现其预期医疗目的的医疗器械” (IMDRF N67).
打破一切的区别:锁定与自适应
传统的设备监管基于一个简单的讨价还价:您证明设备是安全有效的 一次,采用固定设计,然后该设计保持不变。 AI 打破了这个假设。 FDA 2019 年的关键讨论文件准确地划出了界限。一个“锁定”算法是指“每次应用相同的输入时都会提供相同的结果,并且不会随着使用而改变”的算法 — 查找表、决策树、冻结分类器(FDA,2019)。而自适应或持续学习的算法则在部署后不断变化。
这种单一属性——在该领域自我改进的软件——是 AI 需要十年新监管的原因。如果你1月份批准的产品不是6月份医院运行的产品,你到底批准了什么?本报告中的每个框架本质上都是试图回答这个问题,并且 产品总生命周期 (TPLC) ——对设备的整个生命周期进行监督,而不仅仅是许可的那一刻——是共同的回应。
监管机构如何决定审查的难度
IMDRF 的 2014 年风险分类框架设定了当今世界遵循的逻辑:SaMD 应受的审查程度取决于两个因素—— 它提供的信息的重要性(它是告知、驱动还是诊断/治疗?)和 医疗状况的严重程度(不严重、严重或危急)(IMDRF N12)。一个建议拉伸治疗背痛的应用程序和一个标记脑出血的算法不是同一种监管动物,而这个两轴网格就是原因。
世界卫生组织则补充了伦理框架。其2021年报告 人工智能促进健康的伦理与治理 制定六项原则——保护自主权;促进福祉和安全;确保透明度和可解释性;培养责任感和问责制;确保包容性和公平性;并促进响应性、可持续AI (世界卫生组织)。世卫组织于 2023 年提出了监管考虑,并于 2024 年 1 月发布了第一个全球指南,直接针对生成 AI 和大型多模式模型(世界卫生组织).
我们是如何到达这里的
AI 医疗器械的到来并没有取得任何突破;它们在大约十年的时间里逐个国家不断积累。下面的里程碑追溯了 2013 年的定义如何成为五大洲专用的生命周期规则。
AI 医疗器械监管的全球时间表(2013-2026)
在不到十年的时间里,AI SaMD 从 IMDRF 定义转变为五大洲的专用生命周期规则。
| 日期 | 里程碑 |
|---|---|
| 2013年12月 | IMDRF N10 定义“软件作为医疗器械”(SaMD) |
| 2017年1月 | FDAclears Arterys — 第一个云+深度学习临床工具 (510(k)) |
| 2018年4月 | FDA 授权 IDx-DR — 首个自主AI 诊断(De Novo) |
| 2018年5月 | 韩国 MFDS 批准 VUNO Med-BoneAge — 韩国首个 AI 设备 |
| 2018年12月 | 日本 PMDA 批准 EndoBRAIN(III 类)——日本首个 AI SaMD |
| 2019年4月 | FDA 关于AI/ML SaMD 修改的讨论文件(锁定与自适应) |
| 2020 | 中国 NMPA 批准 DeepVessel FFR — 第一个 III 类 AI 设备;日本为 SaMD 推出 IDATEN + DASH |
| 2021 年 1 月 | FDA AI/ML行动计划; 2021 年 6 月 世界卫生组织六项道德原则 |
| 2021 年 10 月 | GMLP — 10 项指导原则 (FDA + 加拿大卫生部 + UK MHRA) |
| 2022 | 沙特 SFDA MDS-G010 — 早期专用 AI 设备指南(被一些人称为第一个“可执行的”); IMDRF N67 ML 术语;巴西 RDC 751/657 |
| 2024 年 8 月 | EU AI 法案生效;世界卫生组织 LMM(生成-AI)指南(2024 年 1 月) |
| 2024 年 12 月 | FDA 最终确定预定变更控制计划 (PCCP) 指南 |
| 2025 年 1 月 | IMDRF N88 GMLP决赛;韩国发布全球首个生成-AI设备指南; FDA AI 生命周期指南草案 |
| 2026 年 2 月 | IMDRF N89 Reliance 手册 |
| 2026 年 4 月 | 加拿大卫生部最终确定了机器学习设备指南;韩国批准首个生成-AI设备 |
资料来源:编译自IMDRF、US FDA、EU、NMPA、PMDA、MFDS、SFDA、ANVISA、加拿大卫生部和世界卫生组织主要文件 — Pure Global,2026 年 6 月。
有几个时刻值得强调。 2017年1月,FDA 批准了 Arterys —— 第一个结合云计算和深度学习的临床工具(美通社)。然后,在 2018年4月,分水岭来了:FDA 授权了 IDx-DR——第一个被允许自主给出诊断、无需医生解读结果的 AI——一款用于初级保健的糖尿病视网膜病变筛查工具在其关键试验中达到了 87.2% 的敏感性和 90.7% 的特异性(npj数字医学)。几个月内,韩国(VUNO Med-BoneAge,2018 年 5 月)和日本(EndoBRAIN,2018 年 12 月)批准了自己的首例;中国紧随其后,于 2020 年推出了 DeepVessel FFR,这是其首个 III 类AI 设备(npj数字医学).
2021 年至 2025 年间,监管框架迎头赶上:2021 年 10 月的三边良好机器学习实践原则,2024 年 8 月生效的 EU AI 法案,FDA 于 2024 年 12 月最终定稿的预定变更控制计划指南,以及——足见前沿发展之快——韩国于 2025 年 1 月发布的全球首个生成式 AI 设备指南(生物世界)。到 2026 年 2 月,IMDRF 已发布全球Reliance 行动手册,帮助监管机构相互采信彼此的工作(IMDRF N89).
数据:有多大、多快、在哪里
间隙曲线急剧向上弯曲
衡量 AI 渗透医学领域的最佳单一标准是 FDA 公开的人工智能医疗器械名单,其最新更新(反映截至 2026 年第一季度的数据)已达到 1,524 项授权(FDA)。为了看到 速度,我们直接分析了底层的FDA 510(k)数据库。已确定的 AI/ML 许可数量从 2010 年代末的少数增加到 2025 年 115 个 - 而且,更能说明问题的是,AI 的份额从 2019 年占所有 510(k) 许可的 0.14% 增至 2025 年的 3.57%,六年内猛增约 25 倍。
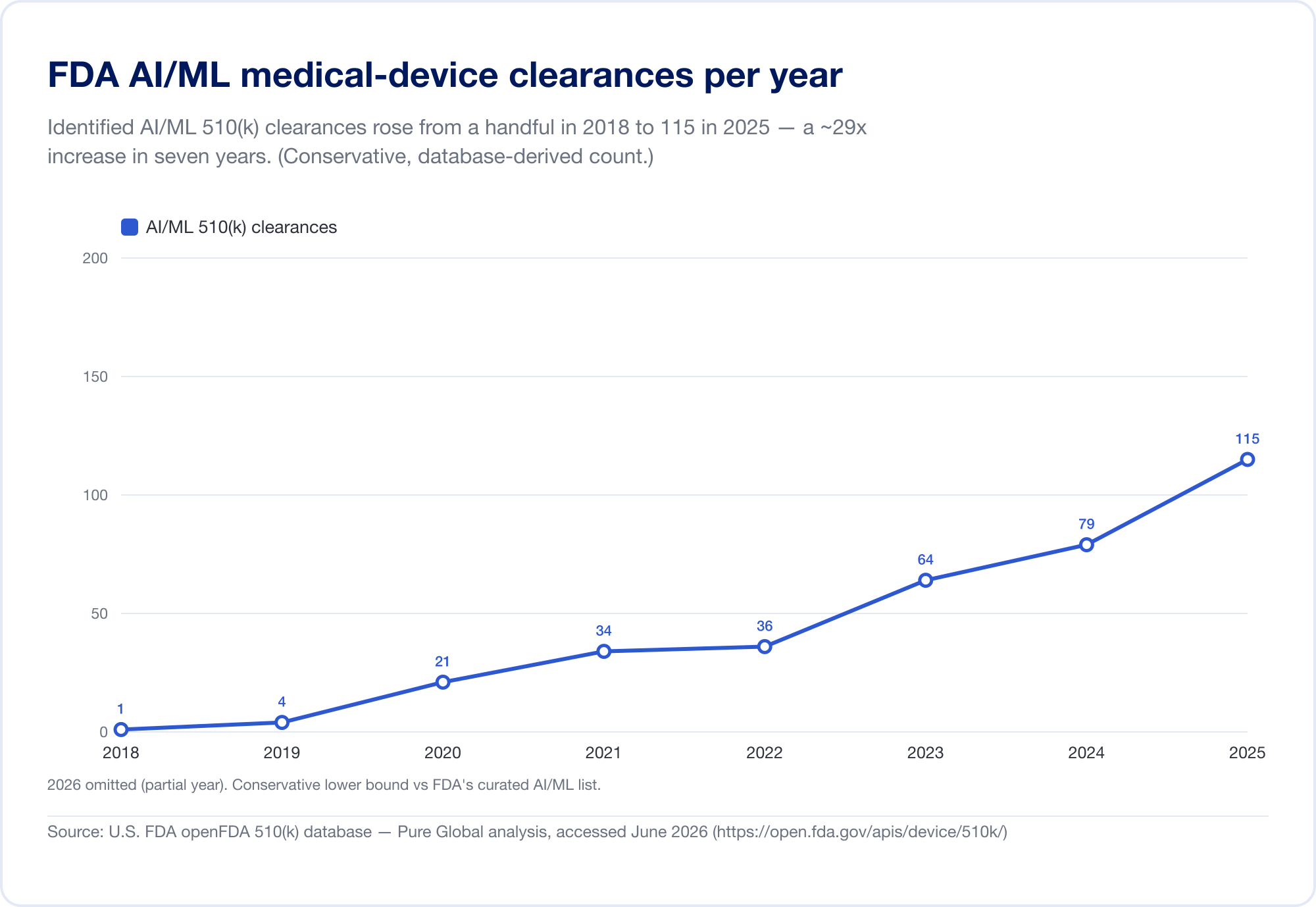
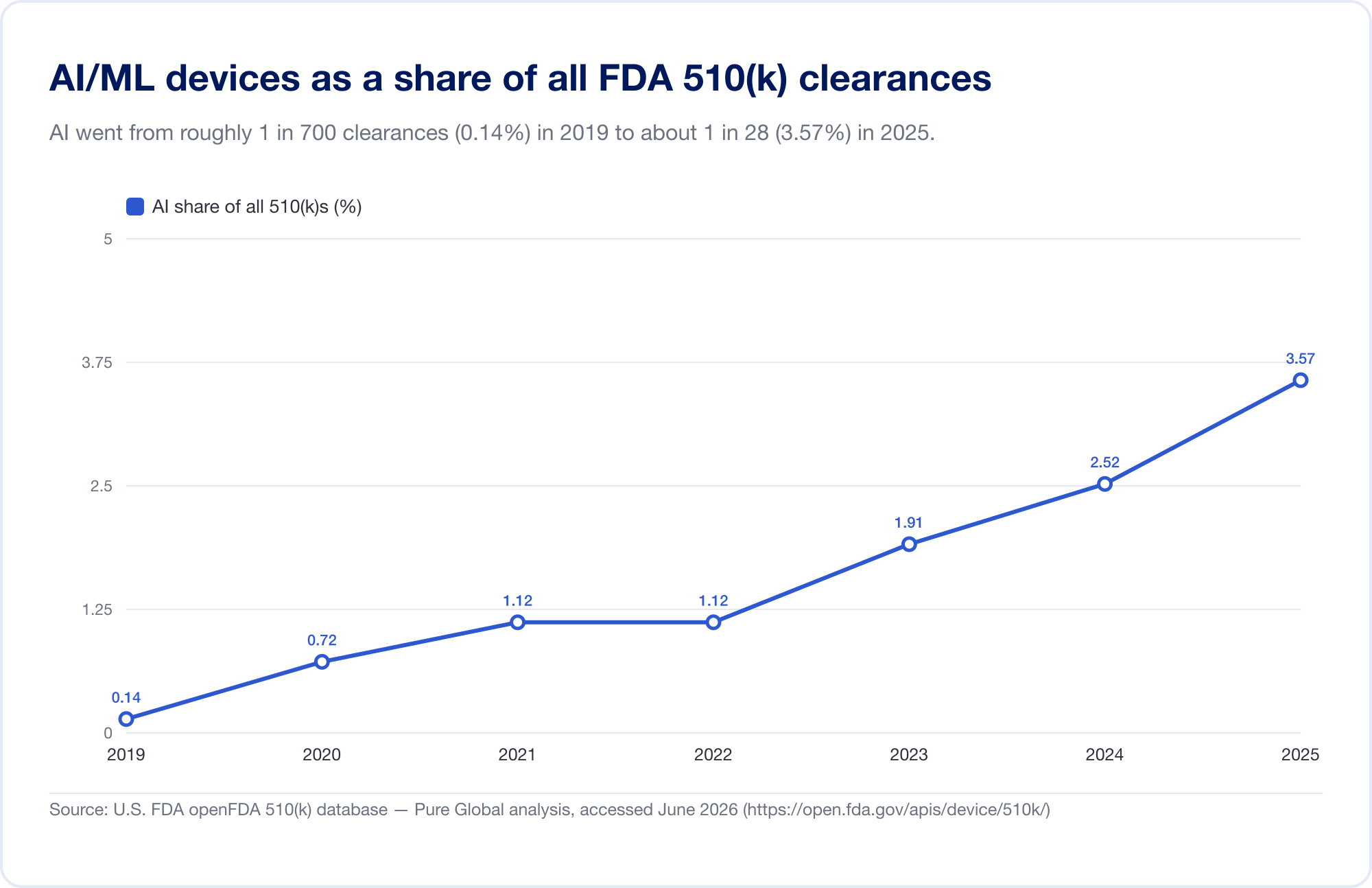
(我们的数据库得出的计数故意保守 - 比 FDA 的策划列表更窄,因为许多 AI 放射学工具位于其文本从未显示“AI”的产品代码下。我们将其用于趋势和地理;FDA 自己的列表是标题总数。来源:openFDA 510(k) 数据库 — Pure Global 分析,6 月2026 年。)
就目前而言,这主要是一个影像故事
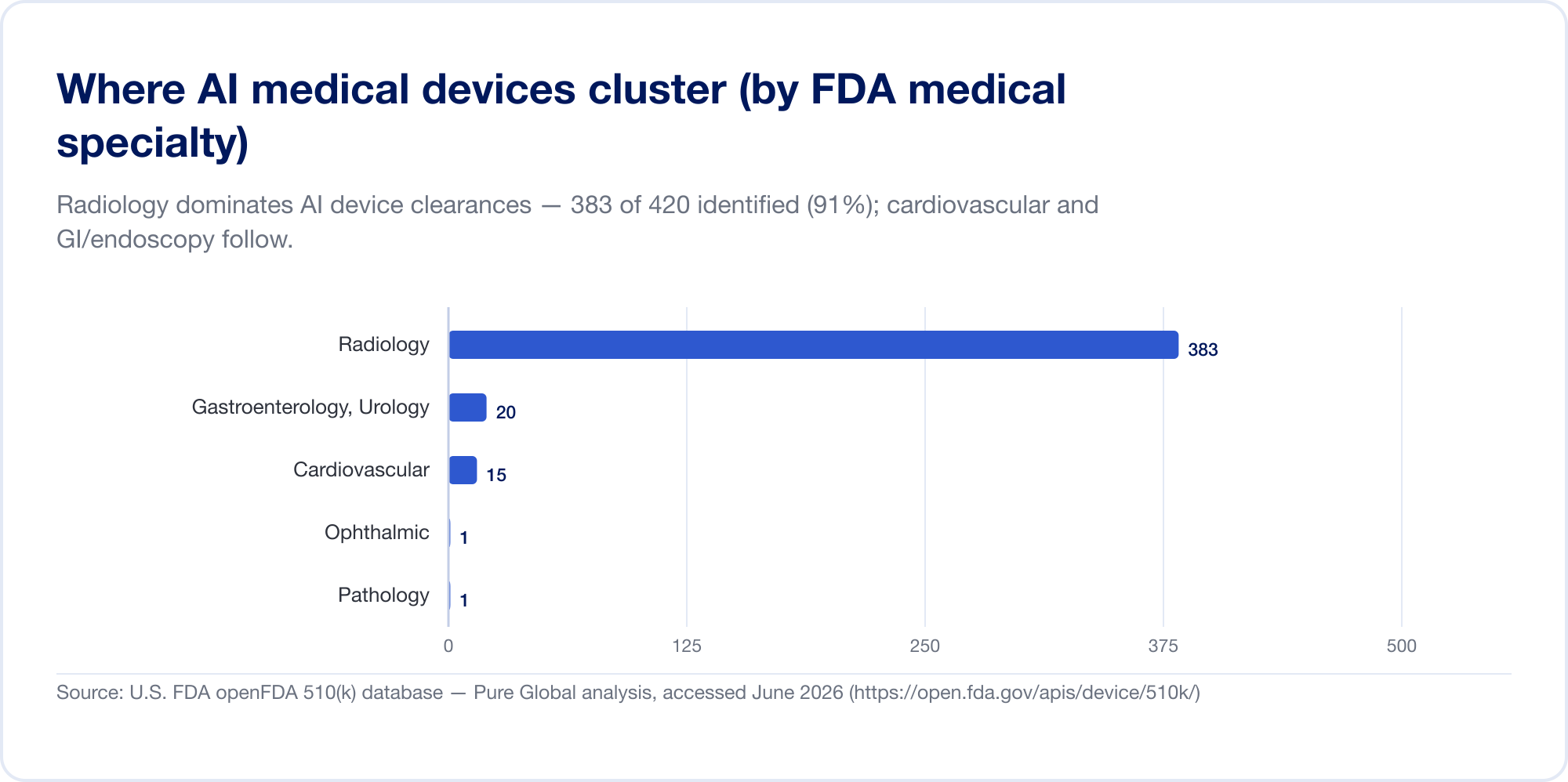
如今,医学领域的AI 主要是放射学领域的AI。在 FDA 的列表中,放射学占所有 AI 授权的约 76%(The Imaging Wire);在我们自己的许可样本中,集中度甚至更高,心血管和胃肠病学(内窥镜检查)以较大差距位居第二和第三。原因是结构性的:成像是数字化的、丰富的、有标签的,510(k) 路径让新算法引用现有算法作为谓词。病理学、心脏病学信号和临床文本模型正在增长,但重心仍然是图像。
创新者无处不在;市场无处不在
这是一个应该重新制定任何商业化计划的事实。 在美国获批的 AI 医疗器械来自至少 16 个国家。在我们对 AI 510(k) 申请人的分析中,美国领先,但韩国、以色列、加拿大、法国、中国、德国、英国、荷兰、台湾和日本各自都提供了有意义的数字。
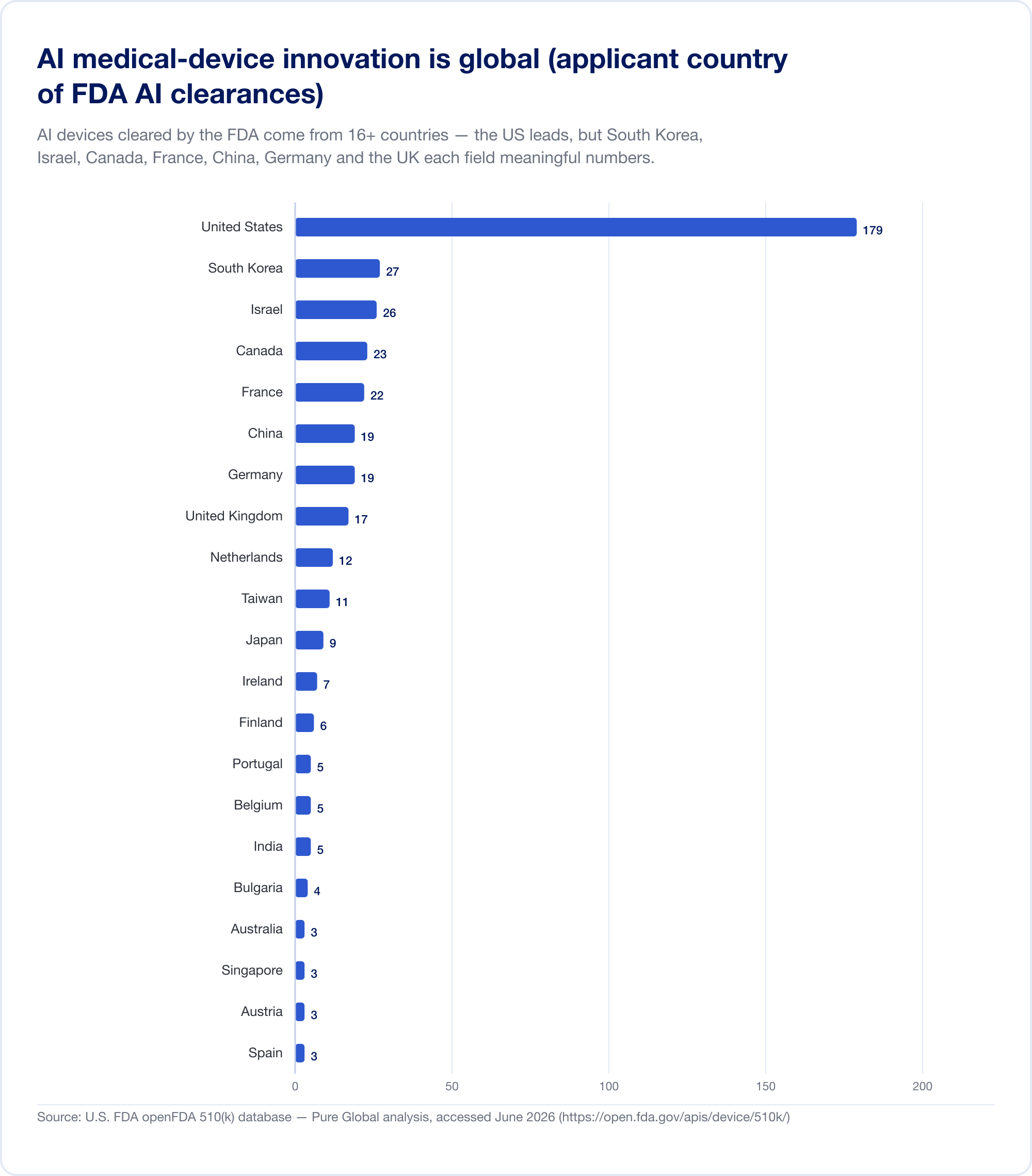
国家产出证实了这一模式。 韩国 2023 年授权(批准和认证合计)64 AI 设备,2024 年授权 108 台,以及 2025 年 153 个 — 增长 41.6%,其中 77.7% 为国产(MFDS). 中国 大致批准了 154 AI医疗器械 到 2025 年中期,其中约 80% 属于风险最高的 III 类(JMIR 医学信息学). 台湾在 2020 年至 2024 年间许可了 166 个 AI/ML 设备(J. Formos. Med. Assoc.)。 日本截至 2025 年 9 月在 PMDA 的名单上有 51 个基于 AI 的 SaMD(全球健康与医学).
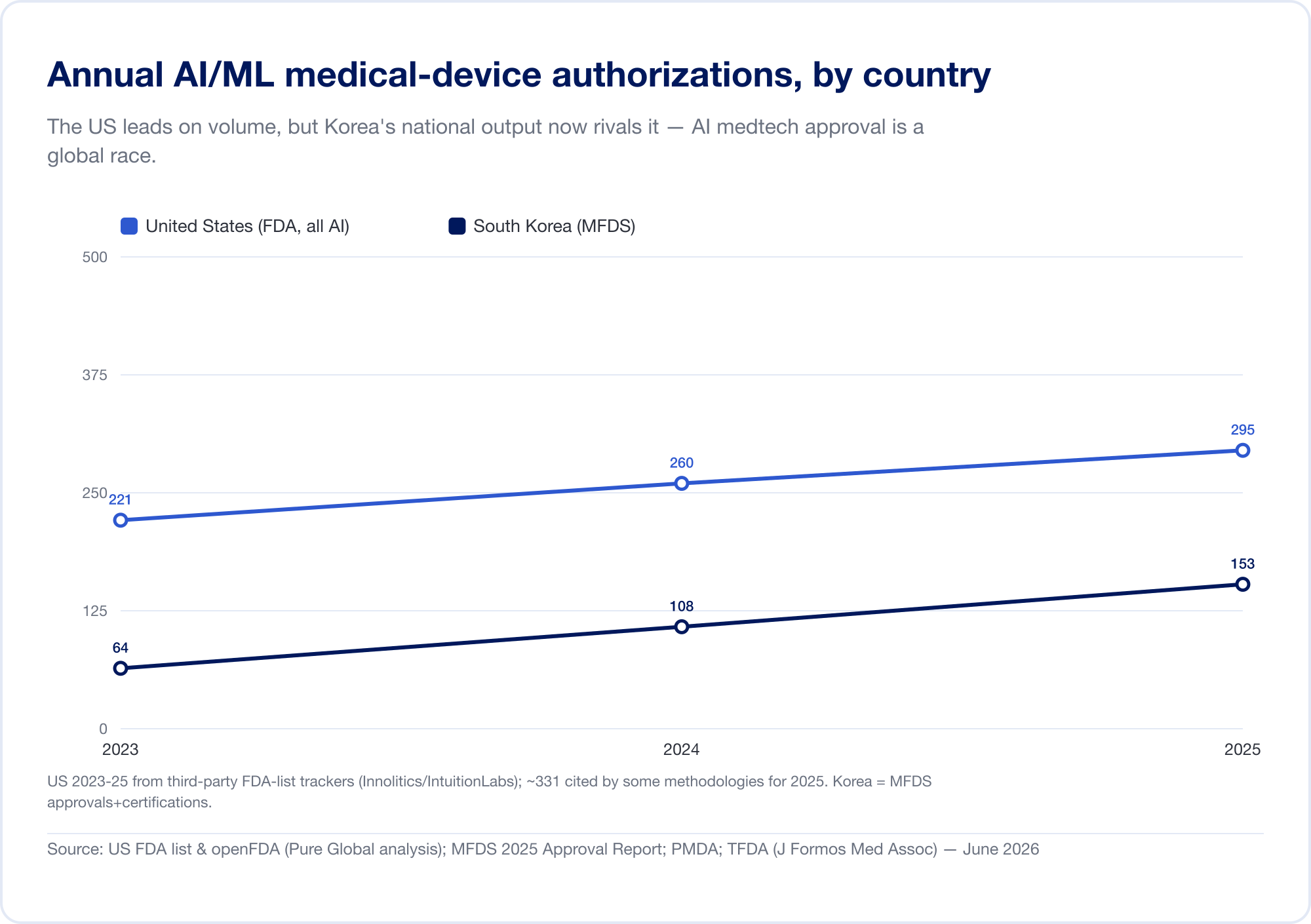
其含义是直接的:在特拉维夫、首尔或上海构建的出色算法必须接触到各个市场中使用不同监管语言的患者。创新是全球性的;批准固然是地方性的。
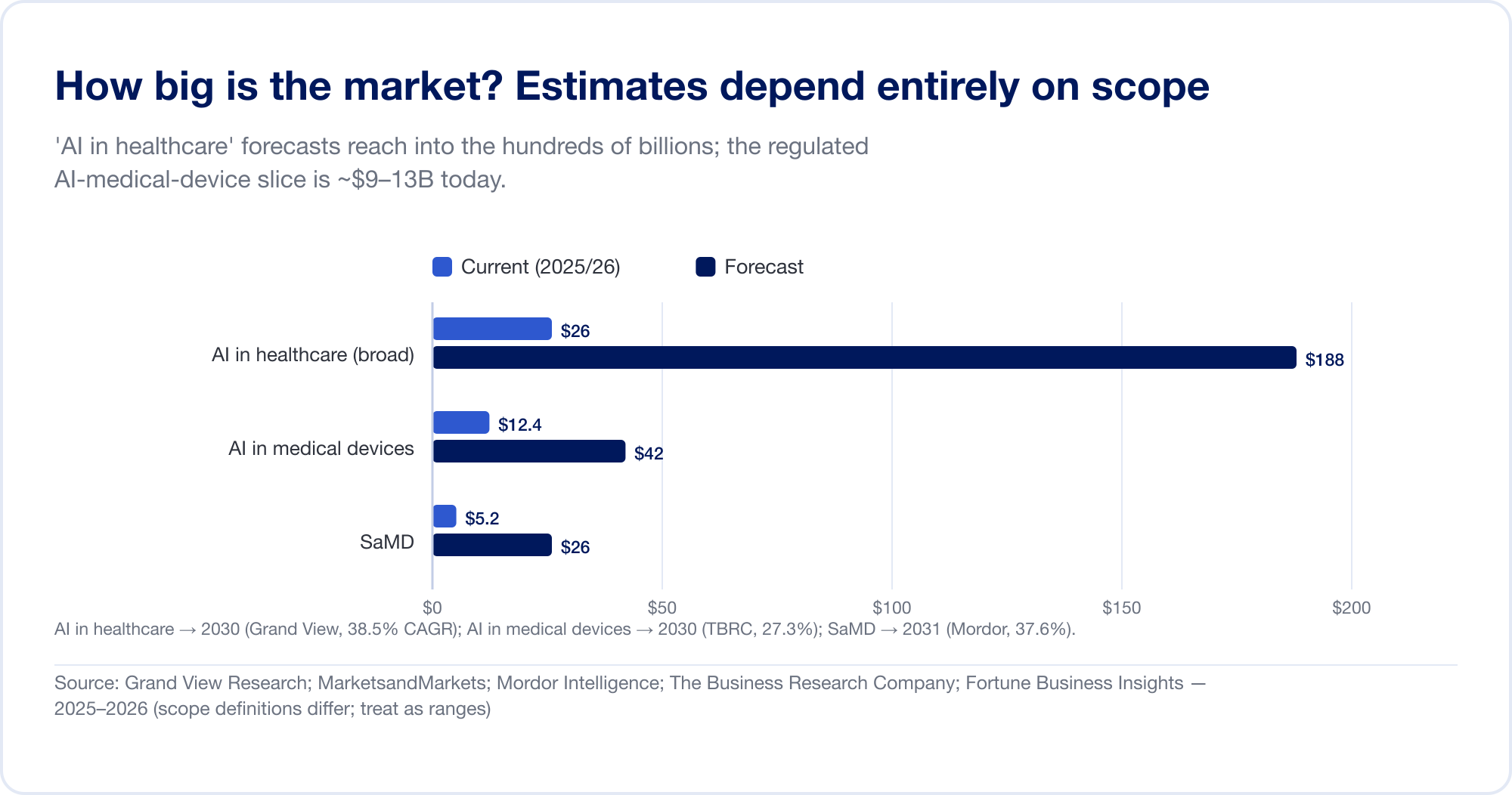
市场有多大?这完全取决于你算什么
对医疗保健领域 AI 的市场规模估计跨越了一个数量级,因为分析师在截然不同的地方划定了界限。广义的 “AI 医疗保健” 类别——捆绑了药物发现、管理自动化、环境抄写员等等——预计在 到 2030 年将达到 1,877 亿美元 年复合增长率为 38.5%(宏景研究),最激进的房屋预计到 2034 年将超过 1 万亿美元(财富商业洞察)。较窄的、受监管的切片—— AI 在医疗器械中 - 更小且更可信:大约 2025年达到124亿美元,到2030年达到424亿美元 (商业研究公司)。纯 SaMD 市场估计约为 50 亿至 250 亿美元,具体取决于来源(魔多情报)。将其中每一项都视为一个范围,而不是事实。
首都讲述了一个更干净的故事。美国数字健康风险投资反弹至 2025 年 142 亿美元(+35%),并且AI启用的初创公司首次占据了大多数—— 占总美元的 54% (岩石健康)。公众AI-SaMD名称正在从增长转向盈利: Tempus AI 2025 财年收入达 13 亿美元(+83%)(Tempus); iRhythm 达到 7.47 亿美元,并迎来第一个 GAAP 盈利季度(iRhythm); HeartFlow 于 2025 年 8 月上市,增长 40% 至 1.76 亿美元(HeartFlow);韩国的 Lunit 增长 53%,其中 92% 的收入来自海外(Lunit)。在非上市公司中,Aidoc 于 2026 年 4 月完成 1.5 亿美元 E 轮融资,融资总额超过 5 亿美元(Aidoc)。
专利竞赛有不同的领导者
知识产权是全球竞争最明显的领域。产权组织里程碑式的研究粗略统计 340,000 份 AI 相关专利申请,其中生命和医学科学为第三大应用领域(世界知识产权组织)。但此后地理格局发生了转变:在 2023 年之前的十年里, 中国提交了 38,210 项生成性-AI 发明——比世界其他国家的总和还多 — 来自美国的 6,276 名(世界知识产权组织)。中国专利局于2024年最后一天发布专门AI专利审查指南(中国国家知识产权局)。医疗 AI 的产品管线正不成比例地由亚洲填补——这使得多市场准入策略对世界上越来越多的开发者而言,成为何时做、而非是否做的问题。
盲点:贸易数据中看不到SaMD
一项分析性警告重新构建了如何思考这个市场。传统的医疗技术市场情报依赖于进口和海关数据——但是 纯SaMD 对它来说是不可见的。通过云下载或应用商店提供的软件不跨越物理边界,不生成 HS 编码海关申报单,也不包含在商品贸易统计中。国际标准制定者直接这么说: “按照现行统计标准,不直接货币化的数据流通常不被视为贸易流” (经合组织-国际货币基金组织-世贸组织),世贸组织自 1998 年起暂停征收电子传输关税(贸发会议)。 AI-嵌入式硬件 — 一台支持 AI 的 CT 扫描仪 — 作为商品移动并显示在数据中; 510(k) 批准的云算法则不然。结果:传统贸易分析系统性地少计了 AI 软件、高估了硬件。 SaMD 全球传播的唯一可靠足迹是其注册记录——这正是本报告所使用的视角。
安全计算
每一次收紧规则的背后都有大量证据表明AI在医学领域可能会以传统设备不会的方式失败。尤其是三项发现重塑了监管机构的思维方式。
隐藏在普通数据中的偏差。 一项具有里程碑意义的新英格兰医学杂志研究发现,脉搏血氧仪——无处不在,并且越来越多地与算法结合使用——漏检危险低血氧水平(隐匿性低氧血症)的比例在黑人患者中为 11.7%,白人患者中为 3.6%,在约 50,000 个配对读数中呈现约三倍的差异(新英格兰医学杂志,2020)。 FDA 于 2021 年 2 月发布了一份安全通讯,并在 2025 年 1 月之前发布了指南草案,要求对肤色进行更多样化的验证(FDA)。这个教训概括起来:一个针对非代表性人群训练的模型可能会将这种偏见悄悄地带入每家部署该模型的医院。
与现实接触后无法继续进行验证。 Epic 脓毒症模型是一种在美国数百家医院部署的专有预测工具,在 38,455 例住院治疗中进行了外部验证,并获得了 曲线下面积为 0.63,远低于供应商声称的 0.76–0.83。它漏掉了 67% 的脓毒症病例,同时对 18% 的患者发出警报(《美国医学会杂志》内科,2021)。一个模型可能已经大规模“投入生产”,但仍然无法像宣传的那样工作。
召回集中在获批后不久。 2025 年约翰霍普金斯大学/耶鲁大学对 FDA 授权的 AI 设备进行的分析发现,43.4% 的 AI 设备召回发生在获批后的前 12 个月内——大约是 510(k) 设备总体召回率的两倍(JAMA 健康论坛,2025)。一项平行研究发现,召回集中在缺乏已发表临床研究的设备中(JAMA Network Open,2025 年)。背景很重要:约 97% 的 AI/ML 设备通过 510(k) 途径获批,该途径不需要前瞻性的人体测试——因此很大程度上取决于上市后的警惕性。
安全思考:监管机构为何收紧AI
偏见、验证差距和早期召回是向生命周期监督转变的证据。
| 寻找 | 图 | 来源 |
|---|---|---|
| 脉搏血氧仪隐匿性低氧血症,黑人与白人患者 | 11.7% 对比 3.6% (~3x) | 新英格兰医学杂志,2020 年 12 月 |
| Epic 脓毒症模型外部 AUC(相对于声称的 0.76–0.83) | 0.63;漏诊了 67% 的脓毒症病例 | JAMA 实习生。医学,2021 年 6 月 |
| AI-设备在清关后 12 个月内召回 | 43.4%(~2x 所有 510(k)s) | JAMA 健康论坛,2025 |
| AI 设备被召回(903 个已研究) | 4.8%,集中在缺乏临床研究的人群 | JAMA 网络公开赛,2025 年 |
| AI 设备通过 510(k) 清除(无需前瞻性测试) | ~97% | FDA 列表分析,2025 年 |
再加上数据集偏移问题 — 随着患者群体、扫描仪或周围编码系统的变化,已部署的模型会悄然退化 — 就构成了整个现代监管体系的基本依据:良好机器学习实践、预定变更控制计划和强制性的真实世界性能监控。监管机构收紧政策并不是因为 AI 不起作用;而是因为它有效——直到它悄然失效。
全球监管地图
这是报告的参考核心:截至2026年中期,主要司法管辖区如何实际分类、审查和监管AISaMD。观看的直线是 分类 (软件属于哪个风险类别)以及 变更控制 (算法更新时会发生什么)。综合比较矩阵遵循区域详细信息。
15 个司法管辖区如何将 AI 作为医疗器械进行监管 (2026)
相同的软件,十五个答案:分类、AI特定指导和变更控制规则因市场而异。
| 管辖范围 | 大多数AI SaMD 降落的地方 | 专用AI/SaMD指导 | 变更控制/自适应-AI机制 | 依赖外国批准 |
|---|---|---|---|---|
| 美国 (FDA) | II 类(510(k)/De Novo) | 是的 — PCCP 决赛 2024 年; 2025 年生命周期草案 | PCCP(预授权变更,无新提交) | 否(自己审查;基于谓词) |
| 欧盟 | IIa+ 类(MDR 规则 11)+ AI 行为高风险 | MDR + AI 法案 + MDCG 2025-6 | 实质性变更 → 公告机构重新审查 + AI 法案 | 否(CE 合格评定) |
| 英国 (MHRA) | IIa+ 级 (UK MDR 2002) | 软件和 AI 更改程序; AI 气闸室 | PCCP 计划(法定文书) | CE 于 2028/2030 年在英国接受 |
| 加拿大(加拿大卫生部) | II-IV 级 | 是 - ML 指南最终版 2026 年 4 月 | PCCP | MDSAP 为QMS;不完全依赖产品 |
| 澳大利亚 (TGA) | IIa-III 级 | AI 2024 年回顾(14 项发现) | 开发中 | 可比海外监管路线 |
| 日本 (PMDA/MHLW) | II-III 级 | DASH for SaMD;SaMD 指导 | IDATEN (PACMP) 预先商定的变更 | 接受国外临床数据; MDSAP |
| 韩国 (MFDS) | 2–3 年级 | 是的——包括。世界第一生成-AI指南 | DMPA 预先批准的变更计划 | 有限公司;自己的评论 |
| 新加坡 (HSA) | B-D 级 | 是 — GL-04-R4 (2025),AI-MD 生命周期 | 变更通知 | 是 - 5 个参考机构(约 98% 删减) |
| 中国 (NMPA) | III类(决策软件) | 是 — CMDE AI 原则 + 分类目录 | 仅在核心算法不变的情况下剥离 | 否(国内代理;型式试验) |
| 印度(CDSCO) | A-D 级 | 仅草稿(2025 年 10 月) | 算法变更协议(提议) | 参考国家/地区批准放宽 C/D 级要求 |
| 台湾 (TFDA) | II-III 级 | 是 - CADe/CADx + PCCP 绘图指南 | PCCP 指南 (2024) | 强调地方绩效评估 |
| 巴西 (ANVISA) | II-IV 级(规则 11) | RDC 657 (SaMD);没有专门的 AI 规则 | 完整变更登记 | 是 — IN 290/2024(III/IV 级,4 个机构) |
| 墨西哥 (COFEPRIS) | I-III 级 | 一般设备规则 | 重新注册 | 是 - 缩写途径 (IMDRF + MDSAP) |
| 沙特阿拉伯 (SFDA) | A-D 级 | 是 - MDS-G010(早期;被称为第一个“可执行”) | 通过 GHAD 更改通知 | 仅支持;需要本地验证 |
| 阿联酋 (EDE) | I-III 级 | 一般设备规则 | 重新注册 | 是 — 识别 CE/FDA |
资料来源:US FDA、EU MDR/AI 法案、MHRA、加拿大卫生部、TGA、PMDA/MHLW、MFDS、HSA、NMPA、CDSCO、 TFDA、ANVISA、COFEPRIS、SFDA、EDE — Pure Global 分析,2026 年 6 月。
美国——基准,也是最繁忙的
美国拥有世界上最活跃的 AI 设备制度,由 FDA 设备和放射健康中心及其数字健康卓越中心管理。没有专门的“AI法规”; 满足器械定义的 AI 功能通过三个途径被作为 SaMD 监管: 510(k) 许可(证明与对照器械“实质等同”)、De Novo 分类(针对没有对照器械的新型中低风险设备),以及 PMA(上市前批准,针对风险最高的 III 类)。绝大多数 AI 设备 — 大约 97% — 通过 510(k) 输入;只有几十个使用了 De Novo,还有少数 PMA(FDA 列表的分析)。具有里程碑意义的 De Novo 是 2018 年的 IDx-DR。
最近的决定性发展是 预定变更控制计划 (PCCP),于 2024 年 12 月最终确定(FDA)。 PCCP 允许制造商预先指定和预先授权一系列未来的模型修改,并且该机构自己的话体现了价值:FDA 审查 PCCP “确保设备的持续安全性和有效性,而无需为实施每次修改而提交额外的营销申请。” 2025 年 1 月,FDA 更进一步,发布了关于支持 AI 的设备软件的整个产品生命周期的全面指南草案。简而言之,美国的态度是:快速、谓词驱动、大量成像,现在围绕生命周期监督进行组织。
欧盟——两种制度叠加在一台设备上
EU 是 AI SaMD 最难的主要市场,因为现在同时适用两种监管制度。首先, 医疗器械法规 (MDR)。其软件分类规则—— 规则11 ——是臭名昭著的。直接阅读: “旨在提供用于诊断或治疗目的决策的信息的软件被归类为 IIa 类,” 当错误决定可能导致死亡或不可逆恶化时升级为 III 类,导致严重恶化时则为 IIb 类(MDR,附件八,通过 EUR-Lex)。根据旧指令,大多数独立软件都自我认证为 I 类,没有第三方参与。规则 11 将几乎所有诊断和治疗 SaMD 推至 IIa 类或以上,从而强制要求公告机构合格评定、ISO 13485 质量管理和临床评估。我们自己扫描EUDAMED数据库发现AI关键字设备集中在IIa类——正是规则11的足迹。
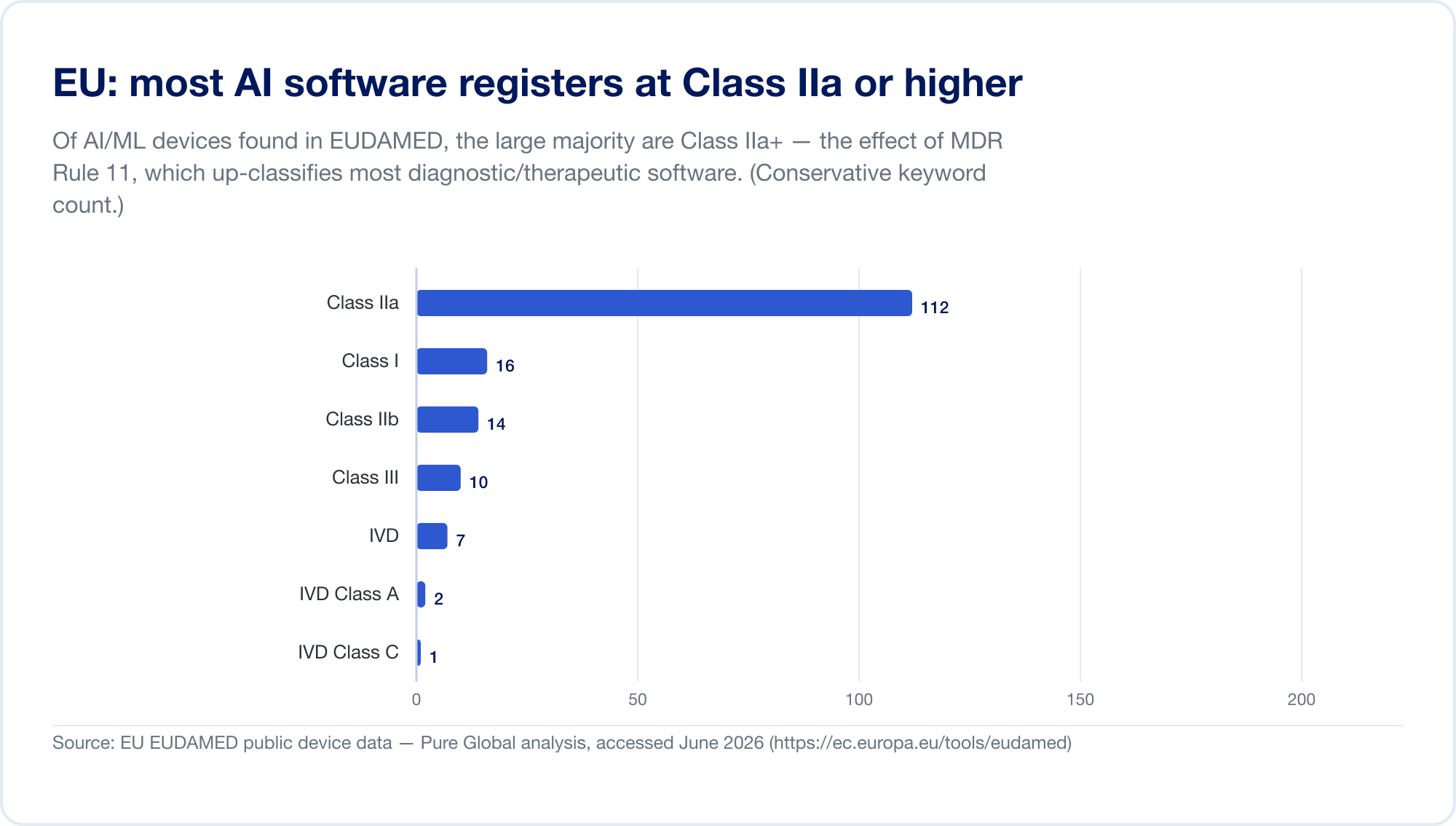
然后,在上面分层, EU AI 法案(第 2024/1689 号条例),自 2024 年 8 月 1 日起生效。根据第 6(1) 条,当 AI 系统本身是(或是其安全组件)已需要第三方合格评定的产品时,即属于“高风险”——这基本上涵盖了所有 IIa+ 类 AI 医疗器械。高风险义务将在 2026 年 8 月 2 日之前逐步实施,MDR/IVDR 设备中嵌入 AI 的日期设置为 2027 年 8 月 2 日 (2025 年 11 月的“数字综合”提案可能会将其推迟到 2028 年——将日期视为移动)(欧盟人工智能法案)。处罚金额高达 3500 万欧元或全球营业额的 7% (第九十九条)。为了澄清重叠,MDCG 和新的欧洲 AI 委员会于 2025 年 6 月发布了联合常见问题解答 MDCG 2025-6 (欧盟委员会).
约束约束是容量。 MDR 公告机构的数量从指令下的大约 80-96 个下降到大约 50,仅在 IVDR 下指定了 ~17–19; MDR 认证现在需要 平均 13-18 个月,大约是 MDR 之前标准的两倍(MedTech 欧洲)。对于 AI SaMD,每台设备都会争夺稀缺的公告机构席位 - 并且在 2027/28 年后还需要 AI 法案合格评定。
英国——分歧、务实
英国脱欧后,MHRA 制定了一条有利于创新的路线。其 软件和 AI 作为医疗器械变更计划 (路线图于 2022 年 10 月发布)涵盖 11 个工作包,该机构已承诺在即将出台的上市前规则中允许 PCCP(MHRA)。其 AI 气闸室 监管沙盒——首个针对 AI 医疗器械的监管沙盒——于 2024 年运行了四个项目试点,并在 2026 年之前运行了七项技术的第二阶段,现已承诺提供多年资金(MHRA)。实际上,英国市场上约 90% 的设备仍然带有CE 标记,英国将接受该标记,直到 2028–2030; MHRA 于 2026 年初就无限期认可 CE 标记进行了咨询(MHRA).
加拿大——第一个最终确定专门的机器学习规则
加拿大卫生部是最早制定相关规定的监管机构之一 最终确定的 针对支持机器学习的医疗器械的专门上市前指南 - 于 2025 年首次定稿,并以修订后的最终形式于 2025 年发布 2026 年 4 月 — 涵盖 II-IV 类,采用 IMDRF 关键术语,并正式引入 PCCP,以便授权的更改不会触发新的许可证修订(加拿大卫生部)。加拿大共同撰写了基础三监管文件——GMLP (2021)、PCCP 原则 (2023) 和透明度原则 (2024)——并自 2019 年起要求 MDSAP 认证。
澳大利亚——早期改革,重新校准AI
澳大利亚的 TGA 早在 2021 年 2 月就改革了其软件规则,推出了低风险健康应用程序,同时对诊断软件进行了升级(具有诊断功能的有源治疗设备移至 III 类)。其2024年咨询, 明确并加强AI的监管,吸引了 600 多名利益相关者,并得出了 14 项主要调查结果,目前正在研究中(TGA)。 TGA 的依赖路线——接受“类似的海外监管机构”——是一个主要的促进剂,如下所述。
日本——为迭代而生
日本将SaMD作为“编程医疗器械”进行监管,并且可以说拥有所有主要市场中最适合AI更新的机制: IDATEN 自 2020 年 9 月起生效,是日本版本的批准后变更管理协议,让制造商预先约定对频繁更新的 AI 进行变更(PMDA)。结合 DASH for SaMD 计划和 SAKIGAKE 优先路径,日本已经为不断演进的软件构建了基础设施——尽管采用率不大:截至 2025 年 9 月,PMDA 列表中只有 51 个基于 AI 的 SaMD。
韩国——行动最快的国家
韩国是其中的佼佼者。除了 2025 年获得 153 项 AI 授权外,它还建立了一个专门的法律框架: 数字医疗产品法 (DMPA)于 2025 年 1 月生效,引入了 PCCP 式变更机制和与 IMDRF 工作项目对齐的数字 QMS(埃默戈)。韩国还于 2025 年 1 月发布了世界上第一个生成式 AI 医疗器械指南,于 2026 年 4 月批准了第一个此类设备,并担任 IMDRF AI/ML 工作组主席。如果您想了解全球 AI 监管的走向,请关注首尔。
新加坡——信赖中心
新加坡 HSA 凭借亚洲最高效的信赖制度,发挥了远超其规模的作用。其软件指导文件 GL-04(修订版 4,2025 年 12 月)明确涵盖整个生命周期中支持机器学习的设备,并且当 AI 模型的性能、输入或人工监督级别发生变化时需要更改通知(HSA)。至关重要的是,HSA 认可五个参考机构(US FDA、EU 公告机构、加拿大卫生部、TGA、日本 MHLW),并且据估计约 98% 的申请可以使用精简途径;已获得两个事先批准的设备可以在短短一个小时内通过“立即”途径进行注册(新加坡卫生部).
中国——幅员辽阔、特色鲜明、要求高
中国NMPA认真对待AI决策支持软件:其 2021 年 CMDE 审查原则和 2021-2022 年分类目录将提供诊断或驱动治疗的软件列为III 类——最高风险等级。中国于 2020 年批准了首个 III 类 AI 设备,到 2025 年中期已达到约 154 个 AI 医疗器械 (JMIR 医学信息学)。在 2025 年 10 月的改革方案中,NMPA 致力于 “简化AI驱动的医疗器械的变更注册要求,其中核心算法保持不变,但算法性能得到优化” — 与美国 PCCP 相比,这是一个真正但狭窄的让步(NMPA)。中国需要国内代理商、本地型式测试,对于许多 III 类设备还需要本地临床数据,这使其成为本报告中要求最高的市场之一。
印度、台湾和亚太其他地区
印度的 CDSCO 于 2025 年 10 月发布了医疗器械软件指南草案,为 AI 更新引入了“算法更改协议”,但它仍然是一个草案,尚未最终确定 AI 规则(CDSCO). 台湾的 相比之下,TFDA 拥有最深入的 AI 指导套件之一 - 专用的 CADe/CADx 技术指南和 PCCP 起草指南 - 并从 2020 年到 2024 年授权了 166 个 AI/ML 设备。在 ASEAN 中,模式是依赖加本地化: 马来西亚、泰国、越南、菲律宾和印度尼西亚 大多数将SaMD视为通用设备并依赖参考国家的批准,越南的快速通道异常广泛(它甚至接受NMPA和MFDS批准)。
拉丁美洲、中东和非洲
巴西的 ANVISA 全盘引入了 EU 的逻辑:其 RDC 751/2022 下的规则 11 与 EU 如出一辙,将决策支持软件置于 II-IV 类,而 RDC 657/2022 是该地区第一个专门针对 SaMD 的决议。外国制造商不能持有巴西注册——本地的巴西注册持有人是强制性且不可豁免的(阿蒂西奥). 墨西哥的 COFEPRIS 在 2025 年对其信赖制度进行了全面改革,将其转变为单一的简化途径,承认所有 IMDRF 和 MDSAP 成员,目标是 30 个工作日。 沙特阿拉伯的 SFDA 发布了 MDS-G010(2022 年 11 月)——世界上最早的专门 AI/ML 医疗器械指南之一,被一些观察家称为第一个可执行的指南(其他人则将其归类为不具约束力)——它独特地规定“制造商应在当地验证在其他司法管辖区开发和批准的基于 AI/ML 的医疗器械”(SFDA)——提醒人们“参考国家的批准”并不总是足够的。阿联酋于 2025 年 1 月在新的阿联酋药品机构下实现了集中设备审批。 南非的 SAHPRA 于 2025 年 9 月发布了第一份 AI 通告,但尚未开始注册设备;而非洲大陆的非洲药品管理局——55 个国家中有 31 个已批准——尚未涵盖器械或 AI。
这是问题的核心:十五个司法管辖区,十五个答案。同样的软件在美国为 II 级,在EU 为 IIa+ 级和“高风险”,在中国为 III 级,在韩国为 2-3 级,在巴西为 II-IV 级——每个软件都有自己的证据、语言、本地持有者和变更控制要求。
费用是多少以及需要多长时间
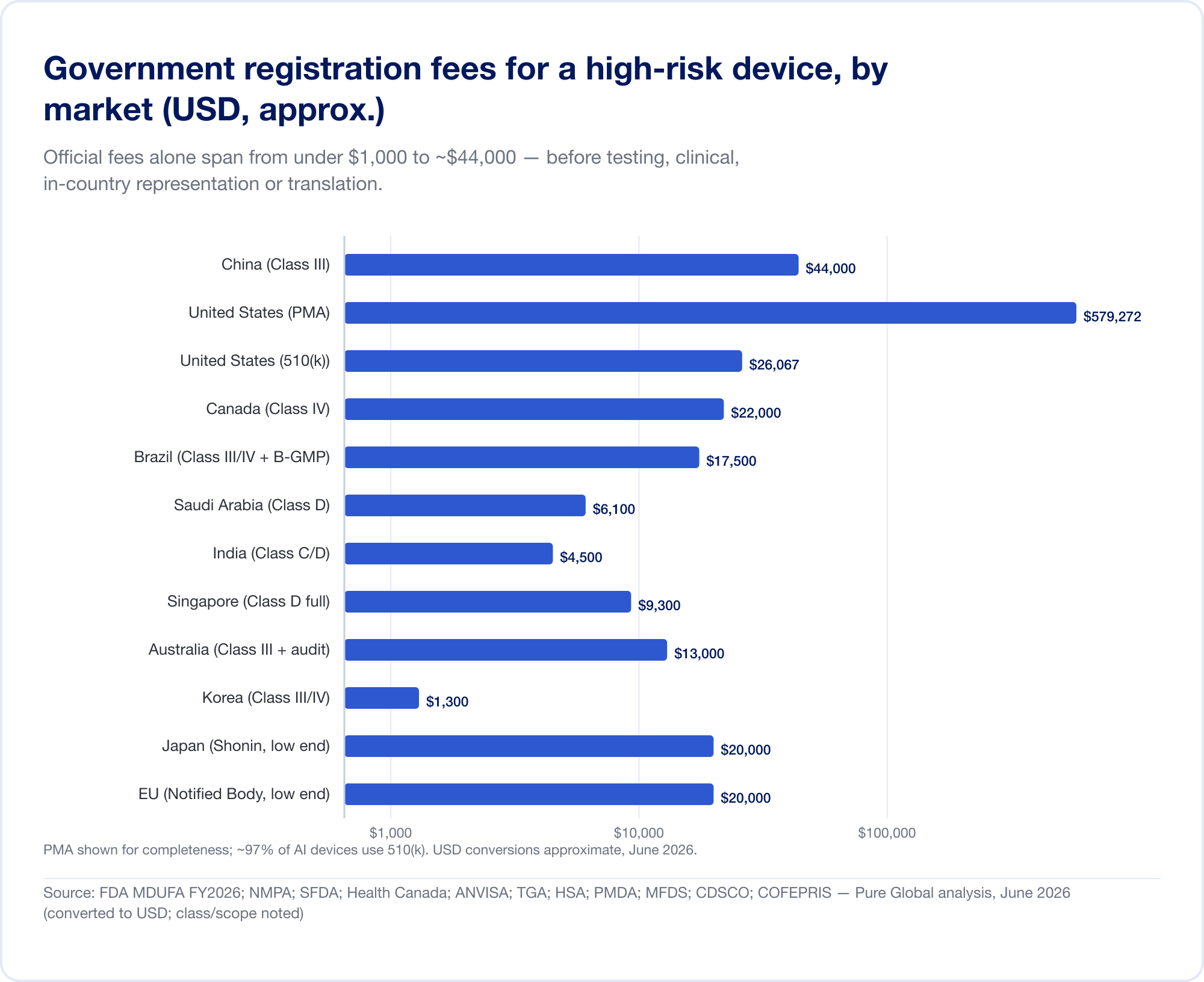
上述分类差异直接转化为金钱和月份。下面的标题数字是 政府费用和现实的时间表 对于风险较高的设备;它们不包括检测、临床证据、翻译和国内代理的巨额费用,这些费用往往使官方费用相形见绌。
官方收费是小数
对于高风险注册,仅政府费用就在 1,000 美元以下到约 44,000 美元之间:
- 美国 — MDUFA 2026 财年费用(在 FDA.gov 上验证):510(k) $26,067(小型企业 6,517 美元);De Novo $173,782;PMA $579,272;加上每年的设施注册费 $11,423 (FDA).
- 中国 — NMPA 注册费大约为 人民币 210,900 元(约 30,000 美元) 对于 II 类和 人民币 308,800 元(约 44,000 美元) 对于 III 类——本报告中最陡的官方费用。
- 巴西 —ANVISA III/IV 级注册费约为 21,000 雷亚尔,另加 B-GMP 认证费 72,804 雷亚尔 对于国际制造商来说。
- 加拿大 — III 级 CAD $14,163, IV 级 CAD $30,713 (2026 年 4 月)。
- 沙特阿拉伯 — SFDA 费用 15,000–23,000沙特里亚尔 按班级。
- 印度 — MD-15进口许可证 每个网站 3,000 美元 + 每个产品 1,500 美元 对于 C/D 类。
- 新加坡、澳大利亚、韩国、日本 — 官方费用相对较低(通常低于 13,000 美元),但证据和审查负担差异很大。
时间是最昂贵的数字
真正的成本是日历。现实的高风险时间表从大约一个月(加拿大,III 类)到 EU 的 24 个月,再到需要本地临床试验时中国的四到五年:
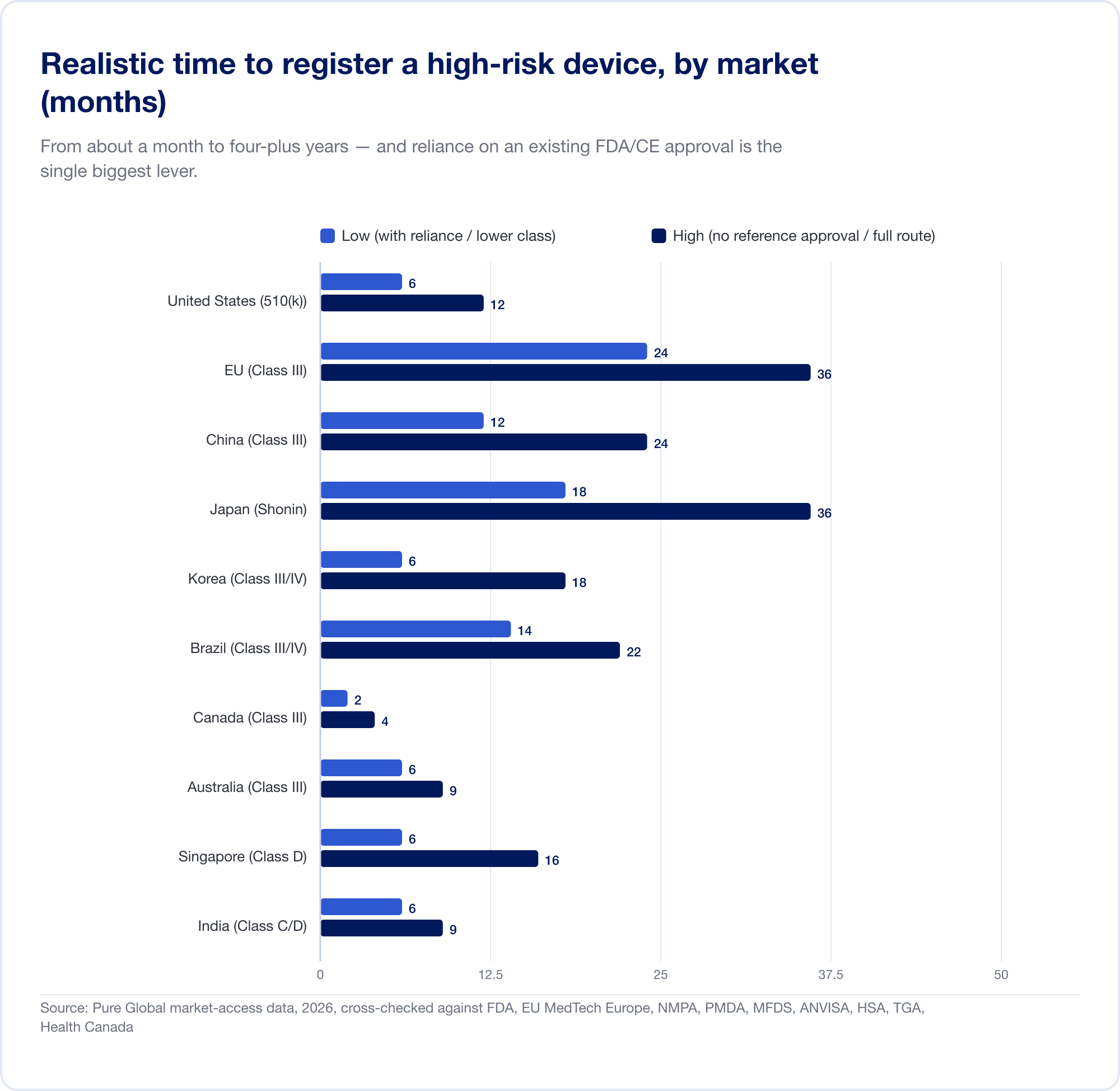
EU 说明了分类如何变成成本。由于规则 11 将过去几乎免费的 I 类自我声明转换为 IIa+ 类公告机构参与,SaMD 的整体 CE 项目通常花费六位数,并需要 13–18 个月才能颁发证书。对于一家拥有以季度为单位的跑道的初创公司来说,这不是一个单项项目,而是一个战略威胁。
杠杆:依赖,以及 AI 特有的扭曲
与这些时间表相对应的是全球市场准入中最强大的工具: 依赖。美国、EU 和中国以外的大多数市场都将依赖于您已经持有的批准。我们自己的市场数据清楚地表明了这种影响——按标准路线在巴西注册需要约 8 个月的高风险 SaMD,在通过 ANVISA 的优化分析途径利用 FDA 或其他参考批准时,可缩短至 6 周。
对于AISaMD,有一个成本表没有显示的特定的、反复出现的变化。当算法更新时——正如 AI 不断更新的那样——问题是该更新是否需要 新的 提交。美国 PCCP 可以让预先指定的更新发布而无需新提交,从而为每个避免的 510(k) 节省 26,067 美元费用加上 90-175 天的审核,避免的 PMA 补充申请则节省更多。 FDA 在 2025 年许可的 AI 设备中,已有超过 10% 嵌入了 PCCP。但这种节省仅限于美国——这是整个报告的关键。
在不同国家将AI注册为医疗器械需要多少钱?
上述政府费用仅为发票的一半。另一半是专业工作——建立档案、进行本地注册、回答当局提出的每个问题,以及提交每次更新和算法更改。这是该行业最不透明的地方:大多数监管咨询公司按小时计费或单独报价每个提交的内容,因此真正的多市场成本只有在变更指令到达后才会出现。
Pure Global 是第一家对每次注册收取单一、固定年费的医疗器械市场准入公司。 来自 每台设备、每个市场、每年 2,000 美元,这笔费用整合了通常按小时计费的服务——国内代表、提交(参考批准)、续订、修改和所有卫生当局通信。没有时间表,也没有每封电子邮件带来的惊喜。
以下是让 Pure Global 作为您的当地代表并按市场进行 AI 医疗器械注册的具体费用。 (AI SaMD 通常属于较高风险类别,因此较高的数字适用于分层市场。)
Pure Global 国内代表 — 每台 AI 设备固定年费
每年每个市场一个透明的号码 - 包括该注册的所有内容。
| 市场 | 本地角色Pure Global提供 | 固定年费(美元) |
|---|---|---|
| 美国 | FDA US 代理 | $1,000 |
| 欧盟 | EU授权代表 | $2,000 |
| 英国 | UK 负责人 (UKRP) | $2,000 |
| 澳大利亚 | TGA 赞助商 | $2,000 |
| 新加坡 | 注册人 | $2,000 · $3,000(C/D 级) |
| 马来西亚 | 授权代表 | $2,000 · $3,000(C/D 级) |
| 泰国 | 授权代表 | $2,000 · $3,000(3/4 级) |
| 印度尼西亚 | 授权代表 | $2,000 |
| 越南 | 市场授权持有人 | $2,000 |
| 香港 | 当地负责人 | $2,000 · $3,000(III/IV 级) |
| 澳门 | 许可证持有者和注册 | $2,000 · $3,000(III 级) |
| 巴西 | 巴西注册持有人 (BRH) | $2,000 · $3,000(III/IV 级) |
| 墨西哥 | 墨西哥注册持有人 | $2,000 · $3,000(II/III 级) |
| 哥伦比亚 | INVIMA代表 | $2,000 · $3,000(IIb/III 类) |
一位 AI SaMD 的国内代表的固定年费;包括提交参考批准、更新、修改和授权信函。来源:Pure Global 主价目表,2026 年(每次注册;多次注册和 3 年合同折扣适用)。
一次性的提交与编制工作——即某个市场需要完整档案时——同样透明公示:美国 510(k) 编制费用为 $15,000–$20,000,EU 技术文档或 CER 项目按类别报价($8,000–$30,000),加拿大注册编制为 $3,000–$25,000(按类别),监管途径判定则为固定 $5,000。每个数字都预先报价,绝不按小时计费。
一个有效的例子——一个 AI 成像算法,四个市场。 拿一个已获得 FDA(II 类)和 CE(IIb 类)标记的 AI 放射学工具,您希望在美国、欧盟、巴西和新加坡保留一年。 Pure Global 的国内代表总数 $1,000 (US) + $2,000 (EU) + $3,000 (巴西) + $3,000 (新加坡) = $9,000 全年 — 固定,包括每次更新、修改和权限交换;仅当市场实际需要新的档案时,您才添加一次性提交工作。可预测性就是关键:当每个市场的规则不同并且您的模型不断变化时,制造商最不需要的就是监管法案也随之变化。
收敛悖论
这是明显的好消息。在十五个司法辖区的拼凑之下,运行着一部强大的协调机器。 IMDRF 统一了定义和良好机器学习实践原则。 医疗器械单一审核计划 (MDSAP) 让单一的质量体系审核同时满足五个监管机构 — 美国、加拿大、巴西、日本和澳大利亚(FDA)。信赖途径正在迅速蔓延:新加坡接受五家参考机构,并通过精简审查来处理约 98% 的申请;巴西、墨西哥、澳大利亚、马来西亚、越南和海湾地区都在某种程度上承认外国批准。 2026 年 2 月,IMDRF 甚至发布了全球Reliance 行动手册,将这一实践编成规范(IMDRF N89).
哪些市场接受哪些外国批准(信赖途径)
FDA 许可或CE 标记是通往数十个市场的万能钥匙,每个市场都有自己的锁。
| 市场 | 公认的参考机构/计划 | 效果 |
|---|---|---|
| 新加坡 (HSA) | US FDA、EU NB,加拿大卫生部,TGA,日本 MHLW | 删节/加急/立即; ~98% 符合资格 |
| 巴西 (ANVISA) | TGA,加拿大卫生部,US FDA,日本 MHLW(III/IV 级) | “优化分析”速度提高约 20–30% |
| 墨西哥 (COFEPRIS) | 所有IMDRF成员+MDSAP参与者 | 简化途径,30 个工作日 |
| 澳大利亚 (TGA) | US FDA,加拿大卫生部,MHLW/PMDA,EU NB,MDSAP | 精简合格评定 |
| 马来西亚 (MDA) | US FDA,加拿大卫生部,TGA,EU CE,PMDA,HSA,泰国FDA | 验证(删节)路线+MDSAP |
| 越南(卫生部) | US FDA、EU、PMDA、TGA、加拿大卫生部、MFDS、NMPA | 异常广泛的 SRA 快速通道 |
| 沙特阿拉伯 (SFDA) | FDA/CE 仅支持 | 仍需要全面的技术文件审查 |
| 阿联酋 (EDE) | CE、US FDA | 基于信赖的注册 |
| MDSAP(一项审核) | US,加拿大、巴西、日本、澳大利亚 | 单一 QMS 审核被所有五个人接受 |
资料来源:HSA、ANVISA、COFEPRIS、TGA、MDA 马来西亚、泰国FDA、越南卫生部、SFDA、EDE — Pure Global 分析,2026 年 6 月。
对于一个 静态的 设备,这是变革性的:一个强有力的认可——通常是 FDA 或 CE——成为以较低的速度和成本打开数十个市场的万能钥匙。这正是运行良好的市场准入计划所要利用的杠杆作用。
这就是悖论。 对于 自适应AI,万能钥匙在最重要的时刻停止工作——改变控制。收敛点在于 装置;分歧在于 AI。考虑寻求更新其算法的相同机器学习模型:
- 在 美国,预授权的 PCCP 可以让其在无需新提交的情况下发布更新。
- 在 EU,“重大”软件更改仍然会触发公告机构重新审查,并且在顶部进行单独的 AI 法案符合性评估。
- 在 中国,只有“核心算法不变”时才容忍更新;真正的再培训意味着全面的变更登记。
- 在 韩国,DMPA 允许预先批准的变更计划——但仅限于预先批准的参数范围内。
正如一项分析直言不讳地说, “FDA 授权的 PCCP 不满足 EU AI 法案义务,反之亦然” (伯克利生命科学公司)。没有 MDSAP 审计,也没有依赖途径可以解决这个问题。 AI 开发商赢得了十个市场的许可,但并没有买到和平;他们买了 十种不同的变更控制义务,每次模型改进时都会触发。对于一项其整个价值主张就是不断变得更好的技术来说,这是对成功的结构性复合税——而对于构建最佳模型的小型、快速发展的团队来说,这种负担最为严重。
下一个前沿:生成AI和基础模型
如果自适应AI给系统带来压力,生成式AI则可能会压垮它。上述所有内容都假设一个模型经过训练,用于单一的、明确的预期用途——检测出血、测量射血分数、标记结节。大型语言模型和多模态基础模型同时从三个方面打破了这一假设: 通用 (一种模型,多种可能的用途), 不确定性 (相同的提示可能会产生不同的答案),并且容易出现 幻觉 (自信、流利、错误)。这些属性都不太适合围绕固定预期用途和“锁定”参考版本构建的框架。
监管机构知道这一点。 世界卫生组织于 2024 年 1 月发布了第一份专门针对大型多模态模型的全球指南,特别警告了伪造的输出、自动化偏差以及验证基于互联网规模数据训练的系统的难度(世界卫生组织)。 FDA 的数字健康咨询委员会于 2024 年 11 月召开首次会议,专门讨论生成式 AI 设备的全产品生命周期挑战(FDA)。而韩国一如既往地率先行动,于 2025 年 1 月发布了世界上第一个生成式 AI 医疗器械指南,并于 2026 年 4 月授权了第一个此类设备。
但指导与清除途径不同。截至 2026 年中期,既定路线 - 510(k)、De Novo、CE 标记 - 仍然假设您可以锁定设备,根据固定标准进行测试,然后冻结。通用临床法学硕士无法完全满足这些先决条件,这就是为什么第一波“医疗保健领域的生成AI”已经进入市场,主要作为回避设备定义的管理工具(环境抄写员、文档辅助工具),而不是作为明确的诊断设备。确实,自主、生成性临床 AI 的监管边界仍在绘制中,而首先绘制该边界的市场(今天的韩国;其他国家将紧随其后)将决定世界其他地区如何复制它们。对于开发商来说,实际的教训是观察界限的设定,并为不断变化的目标设计监管策略,而不仅仅是产品。
市场准入手册
如果问题是碎片化,那么答案就是系统。通过上述模式,出现了一个可重复的剧本,用于将 AI 医疗器械推向世界 - 并将其保留在那里。
1. 在建立证据之前先进行分类。 根据市场和声明,同一产品可以是 II 类或 III 类。绘制目标市场的分类规则 第一,因为它们决定了您需要的临床和技术证据。对最严格的目标市场的主张和证据进行排序可以避免以后重建档案。
2. 赢得主播的认可——然后故意利用信任。 一个FDA 许可或CE 标记的价值远远超过一个市场;凭借此证书,您可以开启新加坡、巴西、墨西哥、澳大利亚、海湾地区及其他地区的精简航线。诀窍在于知道每个目标市场认可哪个锚定批准,并相应地路由档案。沙特阿拉伯要求本地验证,即使是外国批准的AI,这提醒我们,依赖是一张地图,而不是一张毯子。
3. 在强制要求的地方(大多数地方)设立国内代表。 外国制造商不能在巴西 (BRH)、EU(授权代表)、中国(法定代理人)、日本 (MAH/DMAH)、沙特阿拉伯、阿联酋、印度等地拥有自己的注册。每一项都需要当地法人实体进行注册并面对卫生当局。设立三十个这样的实体是不切实际的;将它们外包给单一合作伙伴是规模化变得可行的方式。
4. 将变更控制视为一流的多市场工作流。 这是 AI 特定的规则。建立美国 PCCP 的同时,也要梳理每个市场如何处理同一个更新——并围绕对您业务最重要的、最严格的制度来设计算法的发布节奏。生命周期计划现已成为市场准入计划的一部分。
5. 将其作为一个连接的操作来运行,而不是三十个断开连接的归档。 成本不是任何一次注册;这是协调——翻译、本地持有者、更新日历、变更通知以及数十个独立变化的制度的上市后警惕。
实践中的典型序列。 对于影像 AI 开发者来说,推广通常是这样:先取得 FDA 510(k) 许可或 CE标记 作为锚定批准;同时,向您的本国市场和一个快速信赖市场(新加坡或澳大利亚)提交申请,以积累早期收入;再利用这些批准开通巴西、墨西哥和海湾地区的精简路线;然后进军高投入、高价值的市场——中国和日本——在那里本地测试或临床数据不可避免且周期最长。自始至终,单个变更控制计划都集中维护,并映射到每个市场的更新规则,因此模型改进会在任何必须存在的地方进行归档,而无需在任何地方进行。顺序不是任意的;它有意对现金流、证据重用和稀缺资源市场进行排序。
这正是 Pure Global 的设计初衷:在30 多个市场提供国内代表和 AI 辅助的监管执行,以固定年费交付,而不是行业默认的无上限按小时计费模式。本报告中的数据来自 FDA、EUDAMED、NMPA、PMDA、MFDS、ANVISA 和数十个国家注册机构,以及我们自己的逐个市场成本和时间表数据集,与我们用于对客户的全球推广进行排序的情报相同。如此彻底地绘制迷宫地图的目的是能够引导客户快速穿过它。
结论:需要注意的四件事
AI现在是主流设备类别,数据证明了这一点。 从 2019 年大约七百分之一的 FDA 许可到 2025 年的二十八分之一,仅在美国就授权了 1,500 多台AI 设备,韩国、中国、台湾和日本的国家计划也在快速扩展,AI SaMD 已经从新颖变成了常态。
每个监管机构都对其进行了升级,大多数监管机构都集中在生命周期监管上。 EU 和巴西的规则 11,中国的 III 级,EU AI 法案中的高风险 — 诊断和治疗 AI 被视为严重风险,共同的答案是全产品生命周期控制、GMLP 和预定变更计划。
Reliance 进行一次批准旅行 — AI 部分除外。 协调机制(IMDRF、MDSAP、信赖路线)真正让强大的锚定批准解锁了数十个市场。但自适应-AI 变更控制差异很大,因此随着模型的发展,单一批准并不在任何地方都有效。这种差距是该领域的决定性运营挑战。
竞争优势在于注册机,而不是注册。 当每个市场的规则各不相同并且每年都在变化时,持久的优势属于那些能够在任何地方快速注册并通过每次模型更新保持每个批准有效的人——作为一个互联系统。
与我们交谈
如果您正在构建或扩展 AI 医疗器械,并权衡要进入哪些市场、以什么顺序以及如何在模型改进时保持每个批准有效,这正是我们要解决的问题。欢迎与 Pure Global 交流,制定基于上述数据的市场准入计划——或探索我们的逐个市场注册指南,深入了解任何一个国家。
来源
上文引用的政府和协调机构;关键参考资料分组如下。所有数据均注明日期;市场规模和预测数据是第三方估计,其范围各不相同,应理解为范围。
定义、框架和指导原则
- IMDRF — SaMD:关键定义 (N10,2013); 风险分类 (N12,2014); 支持 ML 的医疗器械:关键术语 (N67,2022); GMLP 指导原则 (N88,2025); Reliance 行动手册 (N89,2026)。 imdrf.org
- US FDA — 软件作为医疗器械 (SaMD); 针对AI/基于机器学习的SaMD 修改的拟议监管框架 (2019); AI/ML行动计划 (2021)。 FDA.gov
- 世界卫生组织 — AI 健康的道德与治理 (2021); AI 健康方面的监管考虑 (2023); LMM指导 (2024); 良好的信赖实践,TRS 1033 附件 10 (2021)。谁.int
美国
- FDA — 人工智能医疗器械 列表(2026 年第一季度更新,1,524 台设备;~76% 放射学); PCCP 最终指导 (2024 年 12 月); 数字健康咨询委员会 关于生成AI(2024 年 11 月); MDUFA 2026 财年费用。 fda.gov · The Imaging Wire(放射学份额分析,2026 年); Innolitics & IntuitionLabs(间隙跟踪器,2025-26)。
欧盟 & UK
- EUR-Lex — 法规 (EU) 2017/745 (MDR),附件 VIII 第 11 条;法规 (EU) 2024/1689(AI 法案),第 6、99、113 条。欧盟委员会 — MDCG 2019-11 Rev.1 和 MDCG 2025-6。 MedTech 欧洲和 Team-NB(公告机构能力)。 MHRA — 软件和 AI 更改程序; AI气闸; CE-识别指导。英国政府网站
加拿大、澳大利亚、日本、韩国
- 加拿大卫生部 — 支持 ML 的医疗器械的上市前指南 (2026 年 4 月)。加拿大网
- TGA — 软件改革(2021); AI 咨询结果(2024-26)。 tga.gov.au
- PMDA/MHLW — SaMD 指导;DASH for SaMD;IDATEN。 pmda.go.jp
- MFDS — 2025 年批准报告(153 个 AI 设备);DMPA;生成式 AI 指南。 mfds.go.kr;bioin.or.kr
亚太地区、拉丁美洲、中东和非洲
- HSA 新加坡 — GL-04-R4;信赖/参考机构。 hsa.gov.sg;新加坡卫生部
- NMPA 中国 — AI 分类和 2025 年 10 月改革。 nmpa.gov.cn · JMIR 医学信息学(154 AIMD,2026)。
- CDSCO India — MD 软件指南草案(2025 年 10 月)。 TFDA 台湾 — CADe/CADx 指南; J.福莫斯。医学。副教授。 (166 个许可证)。
- ANVISA 巴西 — RDC 751/2022(规则 11)、RDC 657/2022、IN 290/2024。 gov.br/anvisa
- COFEPRIS 墨西哥 — 缩写途径(2025)。沙特阿拉伯 SFDA — MDS-G010 (2022)。阿联酋 EDE — 联邦法令 38/2024。 SAHPRA 南非 — AI 通讯 (2025)。
市场、公司、专利、安全
- 市场规模:Grand View Research;市场与市场;商业研究公司;魔多情报; 《财富》商业洞察(2025-26)。
- 资金和公司:Rock Health(2025 年数字健康资金); CB 洞察;公司 2025 财年文件(Tempus AI、iRhythm、HeartFlow、Butterfly Network、Lunit、VUNO); Aidoc(E 系列)。
- 专利:产权组织技术趋势(2019 年)和生成 AI 专利格局(2024 年);中国国家知识产权局。
- 安全性:NEJM(脉搏血氧计偏差,2020); JAMA 内科(史诗脓毒症模型,2021); JAMA 健康论坛和 JAMA Network Open(AI 召回,2025 年); FDA 脉搏血氧计指导。
- 贸易:经合组织-国际货币基金组织-世贸组织 衡量数字贸易 (2021);世贸组织;贸发会议。
- Pure Global 专有市场准入成本和时间表数据集(2026); openFDA,EUDAMED 和 MFDS 数据库分析 — Pure Global,2026 年 6 月。



无论您身在何处,
都欢迎联系我们。
无论您是在寻找更多信息,还是已经准备与我们合作,我们都会在监管流程的每一步提供指导。
联系我们