
IVDR 下有哪些挑战?
IVD 制造商将新的分类系统、预期目的的详细定义以及验证临床证据的需要视为IVDR 下的关键挑战。
IVDR 下有哪些挑战?
根据EU委员会的声明,IVDD和IVDR之间系统的整体结构和方法没有改变。然而,IVD 制造商多年来一直抱怨IVDR 带来的挑战。这些挑战主要源于新的分类系统、针对预期目的的详细定义以及临床证据验证的需要。
这三个要素是如何相互联系的?
IVDD 下没有基于风险的分类系统,IVDs 是根据反映 1998 年最先进技术和知识的肯定列表进行分类的。由于该系统不够充分,EU 决定与全球协调工作组 (GHTF) 在 2008 年推荐的分类系统保持一致,该系统已在许多国家使用。基于 GHTF 的新系统将 IVDs 分为四个基于风险的类别,引入风险分类和实施规则来确定 IVD 所需的监管合格评估级别。
作为其中的一部分,分类规则现在与制造商定义的预期目的和声明相关联。例如,与 IVDs 相比,用于诊断癌症或血型的设备被归类为更高的风险类别,其中诊断不会对患者健康产生严重影响。
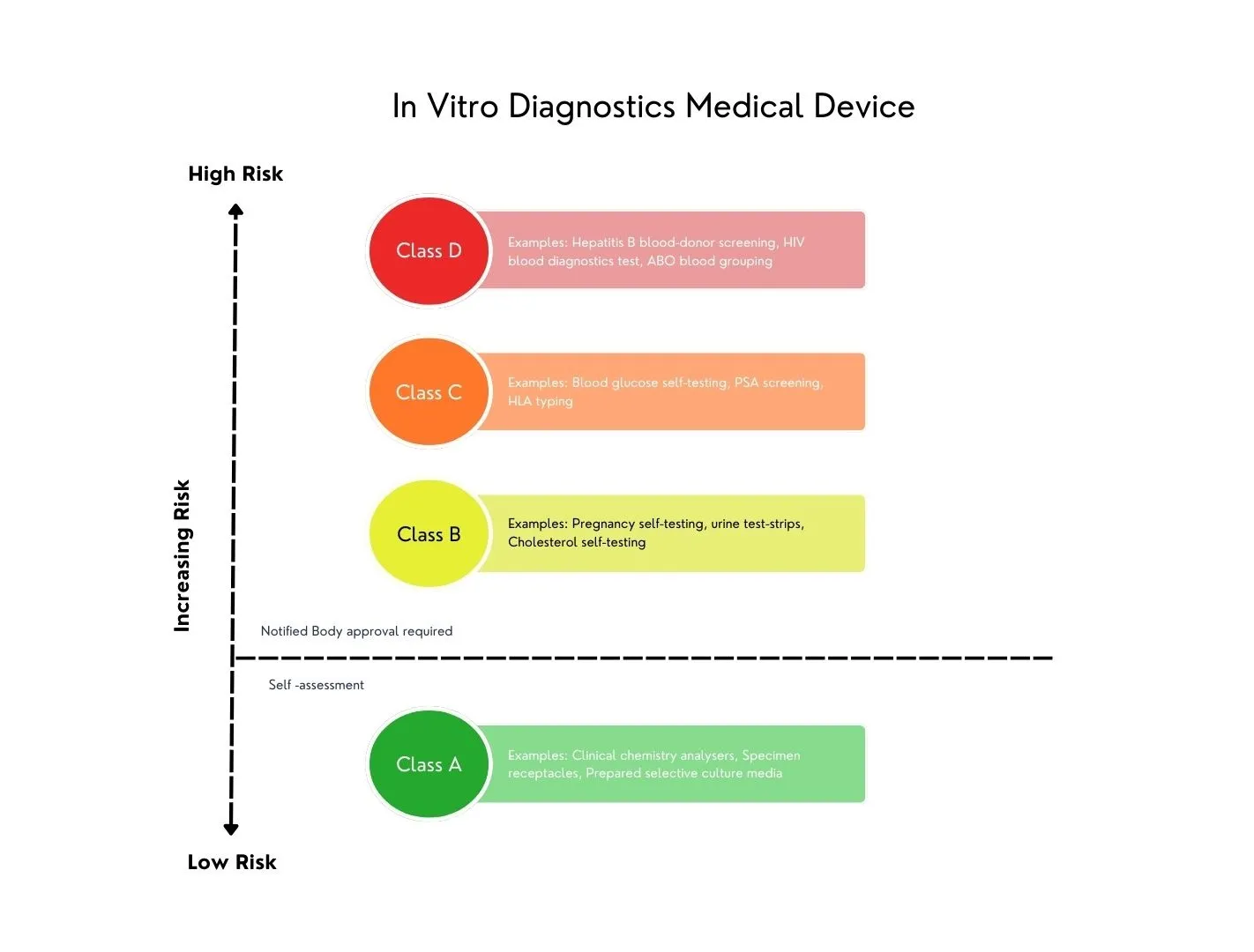
这种转变意味着预期目的必须不仅仅是反映分析测量。它必须与制造商定义的声明和指示一致。此外,这些声明必须作为临床证据的一部分进行验证,其中包括科学有效性、分析性能和临床性能,通过性能评估流程(单独的计划和报告)。
这一过程的主要挑战之一是从纯粹的分析预期目的(例如“打算在 EDTA 血浆中测量”)转变为包括目标群体、适应症和使用场景的更全面的定义。例如,“打算在自动分析仪[设备名称]上测量[年龄范围]的人类患者的 EDTA 血浆,以进行癌症筛查。”这需要高度的细节和清晰度。
为什么这会影响制造商?
IVDs 现在需要通过性能数据对所有声明的预期目的进行明确验证和记录,如果临床医生仅将其用于指定目的,这可能会限制销售。此外,对于组合设备,例如自动分析仪上使用的IVD套件,必须明确说明和验证组合使用。如果制造商已经有足够的数据来支持这些说法,那么从 IVDD 到 IVDR 的转变可能不是一个重大挑战。然而,缺乏这些数据可能会导致销售和监管部门之间就预期用途声明中应包含哪些内容进行大量讨论。这还需要大量工作来验证这些声明,使其与分类和临床证据保持一致,其中可能包括临床性能研究,以确认它们对目标群体和预期使用场景的用途。
总之,IVDs分类系统的变化对预期目的和临床证据产生了重大影响。对于医疗器械来说,类似的基于风险的分类系统已经成为MDD的一部分,这就是为什么医疗器械制造商已经习惯了它,而IVD制造商尚未习惯它。这些挑战继续影响着许多 IVD 制造商,并且通常只有当他们的数据首次由独立的公告机构审查和评估时,这些挑战才会变得明显。与许多其他事情一样,良好的规划和意识是满足监管要求的关键。通过适当的准备,IVDR 下的 CE 标记并不像看起来那么困难。
当IVD 制造商应对IVDR 合规性的复杂性时,专家指导和先进AI 工具的使用可以产生重大影响,从而节省时间和金钱。请联系 Pure Global,我们将帮助您克服监管挑战,并确保您的产品满足所有必要的 IVDR 要求。
本地化 要点 摘要
本文 面向 医疗器械 和 IVD 制造商,梳理 法规 策略、市场 准入、质量 管理、技术 文件、临床 证据、本地 代表、注册 时限、上市后 监督、风险 管理 和 团队 分工 的 关键 内容。
- 提前 规划 可以 降低 申请 延误、资料 缺口、补充 问题、认证 机构 沟通 和 市场 中断 风险。
- 企业 需要 明确 每个 市场 的 分类、法规 路径、文件 要求、标签 翻译、注册 持有人 和 合规 责任。
- 内部 团队 与 外部 专家 的 分工 应 结合 产品 风险、目标 市场、预算、时间 表 和 长期 维护 能力 来 决定。
- 法规 要求 会 持续 更新,获批 后 仍然 需要 监测、报告、续证、变更 管理、纠正 措施 和 证据 维护。
- Pure Global 结合 多市场 实务 经验、本地 网络、AI 辅助 工具 和 法规 专业 知识,支持 企业 更高效 地 进入 全球 市场。



无论您身在何处,
都欢迎联系我们。
无论您是在寻找更多信息,还是已经准备与我们合作,我们都会在监管流程的每一步提供指导。
联系我们