
Was sind die Herausforderungen unter IVDR?
IVD-Hersteller nennen das neue Klassifizierungssystem, detaillierte Definitionen für beabsichtigte Zwecke und die Notwendigkeit der Überprüfung klinischer Beweise als größte Herausforderungen im Rahmen des IVDR.
Was sind die Herausforderungen unter IVDR?
Laut Aussagen der EU-Kommission haben sich die Gesamtstruktur und der Ansatz des Systems zwischen IVDD und IVDR nicht geändert. Allerdings beschweren sich IVD-Hersteller seit Jahren über die Herausforderungen, die IVDR mit sich bringt. Diese Herausforderungen ergeben sich hauptsächlich aus dem neuen Klassifizierungssystem, detaillierten Definitionen für beabsichtigte Zwecke und der Notwendigkeit der Überprüfung klinischer Beweise.
Wie hängen diese 3 Elemente miteinander zusammen?
Unter IVDD gab es kein risikobasiertes Klassifizierungssystem, und IVDs wurden auf der Grundlage einer Positivliste klassifiziert, die den neuesten Stand der Technik und des Wissens von 1998 widerspiegelte. Da dieses System unzureichend wurde, beschloss EU, sich an das von der Global Harmonization Task Force (GHTF) im Jahr 2008 empfohlene Klassifizierungssystem anzupassen, das bereits in vielen Ländern verwendet wird. Das neue GHTF-basierte System klassifiziert IVDs in vier risikobasierte Klassen und führt Risikoklassifizierungs- und Implementierungsregeln ein, um den Grad der regulatorischen Konformitätsbewertung zu bestimmen, die für ein IVD erforderlich ist.
Dabei werden Klassifizierungsregeln nun an den vom Hersteller definierten Verwendungszweck und die Ansprüche geknüpft. Beispielsweise wird ein Gerät zur Diagnose von Krebs oder zur Blutgruppenbestimmung in eine höhere Risikoklasse eingestuft als IVDs, wenn die Diagnose keine schwerwiegenden Auswirkungen auf die Gesundheit des Patienten hat.
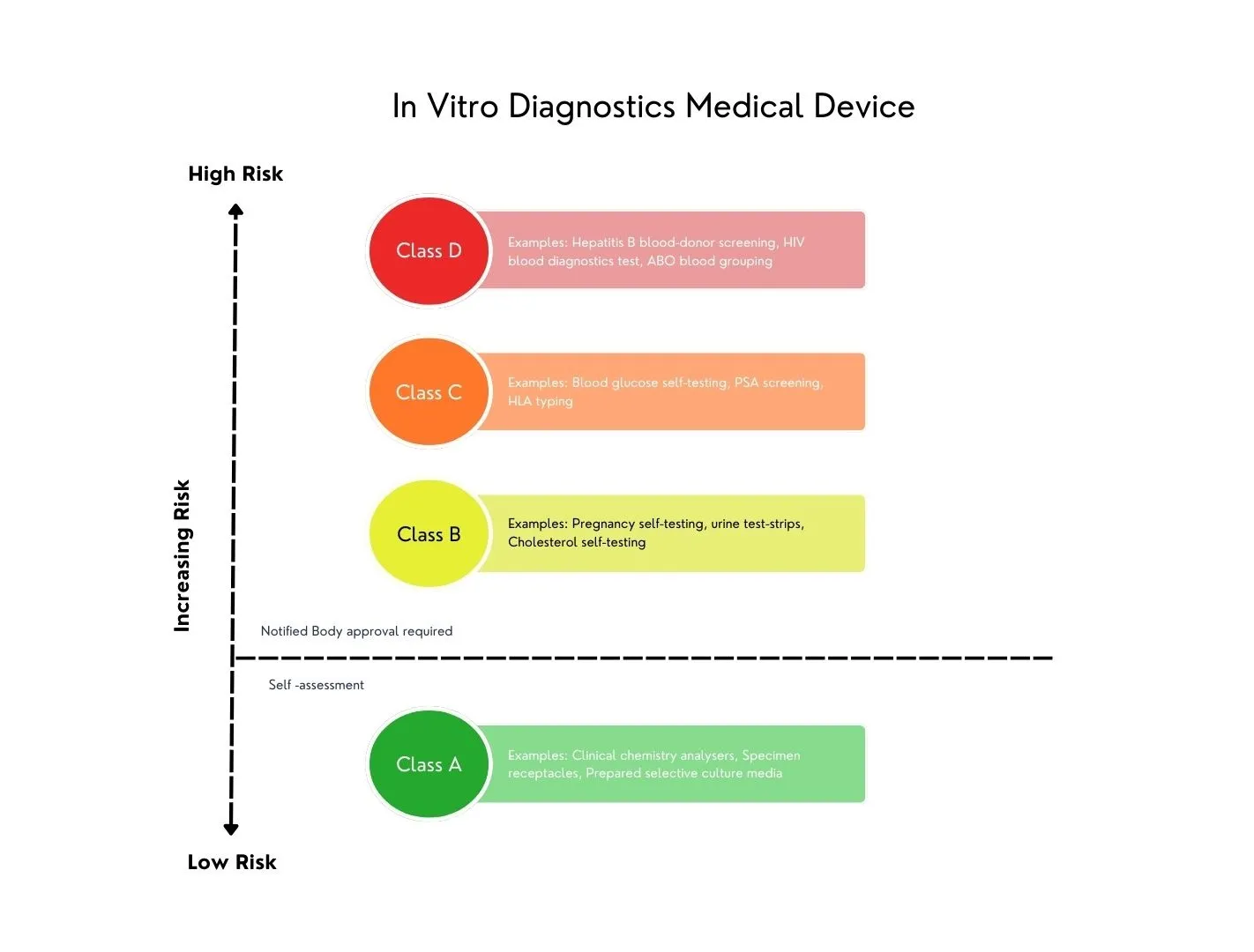
Diese Verschiebung bedeutet, dass der beabsichtigte Zweck über die bloße Wiedergabe der analytischen Messung hinausgehen muss. Es muss den vom Hersteller definierten Angaben und Angaben entsprechen. Darüber hinaus müssen diese Behauptungen als Teil der klinischen Evidenz, die wissenschaftliche Validität, analytische Leistung und klinische Leistung umfasst, durch einen Leistungsbewertungsprozess (separater Plan und Bericht) verifiziert werden.
Eine der größten Herausforderungen in diesem Prozess ist der Übergang von einer rein analytischen Zweckbestimmung, etwa „zur Messung in EDTA-Plasma vorgesehen“, zu einer umfassenderen Definition, die Zielgruppe, Indikation und Anwendungsszenario einbezieht. Zum Beispiel „zur Messung in EDTA-Plasma auf dem automatischen Analysegerät [Gerätename] für menschliche Patienten im Alter von [Altersgruppe] zur Krebsvorsorge vorgesehen.“ Dies erfordert ein hohes Maß an Detailliertheit und Klarheit.
Warum wirkt sich dies auf die Hersteller aus?
IVDs verlangen nun eine klare Überprüfung und Dokumentation aller Ansprüche in Bezug auf ihren beabsichtigten Zweck durch Leistungsdaten, was den Verkauf einschränken kann, wenn Ärzte sie nur für den angegebenen Zweck verwenden. Darüber hinaus muss bei kombinierten Geräten, wie z. B. IVD-Kits, die auf automatischen Analysegeräten verwendet werden, die kombinierte Verwendung klar angegeben und überprüft werden. Wenn Hersteller bereits über ausreichende Daten zur Untermauerung dieser Behauptungen verfügen, stellt der Übergang von IVDD zu IVDR möglicherweise keine große Herausforderung dar. Das Fehlen dieser Daten könnte jedoch zu erheblichen Diskussionen zwischen Vertriebs- und Regulierungsabteilungen darüber führen, was in den Zweckbestimmungen enthalten sein sollte. Dies wird auch umfangreiche Arbeiten erfordern, um die Angaben zu überprüfen und sie mit Klassifizierung und klinischen Beweisen in Einklang zu bringen. Dazu können klinische Leistungsstudien gehören, um ihre Verwendung für die Zielgruppe und das beabsichtigte Verwendungsszenario zu bestätigen.
Zusammenfassend lässt sich sagen, dass die Änderungen im Klassifizierungssystem für IVDs erhebliche Auswirkungen auf den beabsichtigten Zweck und die klinische Evidenz haben. Für Medizinprodukte ist ein ähnliches risikobasiertes Klassifizierungssystem Teil des MDD, weshalb Medizingerätehersteller daran gewöhnt sind, während IVD-Hersteller noch nicht daran gewöhnt sind. Diese Herausforderungen betreffen weiterhin viele IVD-Hersteller und werden oft erst sichtbar, wenn ihre Daten zum ersten Mal von einer unabhängigen benannten Stelle überprüft und bewertet werden. Wie bei vielen anderen Dingen sind gute Planung und Bewusstsein der Schlüssel zur Erfüllung regulatorischer Anforderungen. Bei richtiger Vorbereitung ist die CE-Markierung unter IVDR nicht so schwierig, wie es scheint.
Während sich IVD-Hersteller mit der Komplexität der IVDR-Compliance auseinandersetzen, können fachkundige Anleitung und der Einsatz fortschrittlicher AI-Tools einen erheblichen Unterschied machen und sowohl Zeit als auch Geld sparen. Kontaktieren Sie Pure Global, um Unterstützung bei der Bewältigung regulatorischer Herausforderungen zu erhalten und sicherzustellen, dass Ihr Produkt alle erforderlichen IVDR-Anforderungen erfüllt.



Sprechen wir,
wo immer Sie sind.
Ob Sie weitere Informationen suchen oder bereit zur Zusammenarbeit sind: Wir begleiten Sie durch jeden Schritt des regulatorischen Prozesses.
Kontakt