
IVDR下的临床证据:IVD制造商的“适当水平”是什么?
遵守 IVDR 要求对临床证据质量采取严格的方法。在本文中,Oliver Eikenberg 博士解释了绩效评估流程和文档的重要性,以确保 EU IVDR 合规性有足够的临床证据。
临床证据并不是体外诊断设备法规 (EU IVDR 2017/746) 下的新要求;然而,与IVDD相比,它的定义更加清晰。 IVDR第56条和附件十三将临床证据定义为一个持续的IVDs 绩效评估 (PE)过程,其中包括科学有效性、分析性能和临床性能。简而言之,临床证据包括性能评估的数据和结果,以证明您的 IVD 是安全的并且按照其预期目的运行。
考虑到IVDR针对预期目的扩展了标准,并引入了更严格的总体安全方法,许多IVDR制造商想知道他们需要走多远才能确保提供足够的临床证据,特别是随着IVDR过渡期限的临近。
然而,许多 IVD 制造商仍然低估了仔细的 PE 流程的必要性,以及向公告机构 (NB) 提供明确评估和分析的 PE 数据以及充足信息的好处。由于临床证据是安全有效器械的总体决策标准,因此完整的 PE 文档、流程和基于风险的准确数据分析是通过 NB 审查的关键。
IVDR下的绩效评估是什么?
绩效评估 (PE) 是支持您的临床证据的机制。它应该是一个结构良好、透明、迭代和连续的过程,通过该过程对数据进行评估和分析,以证明设备针对其预期目的的科学有效性、分析性能和临床性能。它必须是制造商 QMS 的一部分,并且发生在包括上市后在内的整个生命周期中。
与许多其他流程一样,PE 从规划开始。绩效评估计划 (PEP) 必须定义数据收集方法、时间表、验收标准和人员职责。然后根据PEP进行PE测试的PE数据收集。根据定义的数据分析收集和评估(评估)可用的科学或PE数据,其结论将告知绩效评估报告(PER)。上市后活动包括对最新技术(例如标准)的持续监测和评估,以及上市后性能跟踪 (PMPF) 活动,这些活动记录在制造商的 PMPF 报告和定期安全更新报告(PSUR)中(如适用)。风险管理是所有这些活动的核心,应包含在每个步骤中。
MDCG 2022-2 用简化的图表说明了 PE 流程:
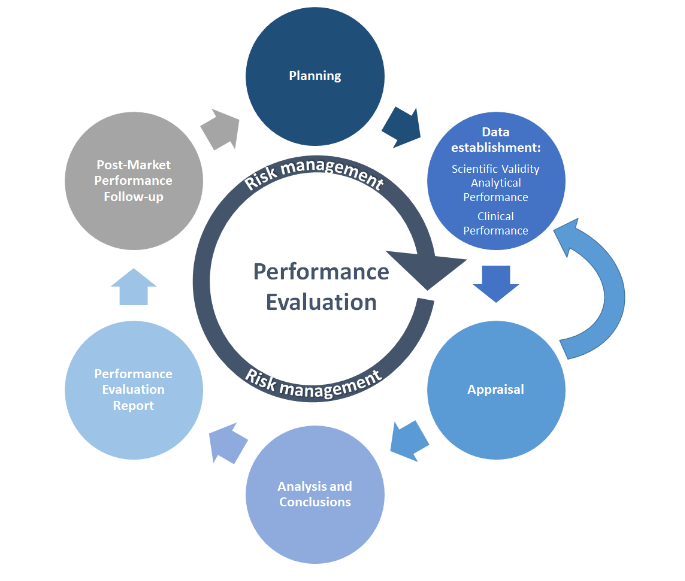
来源:MDCG 2022-2
如前所述,IVDs 的临床证据可以通过收集科学有效性、分析性能和临床性能的 PE 数据来建立。证据的收集和记录应达到允许对设备安全性以及设备在按制造商预期使用时是否达到预期临床益处进行合格技术文件评估(由公告机构进行的 TDA)的水平。这并不一定意味着必须进行临床性能研究。有时,通过合格的评估人员进行科学有效性评估以及使用足够数量的临床样本进行分析性 PE 测试就足够了。
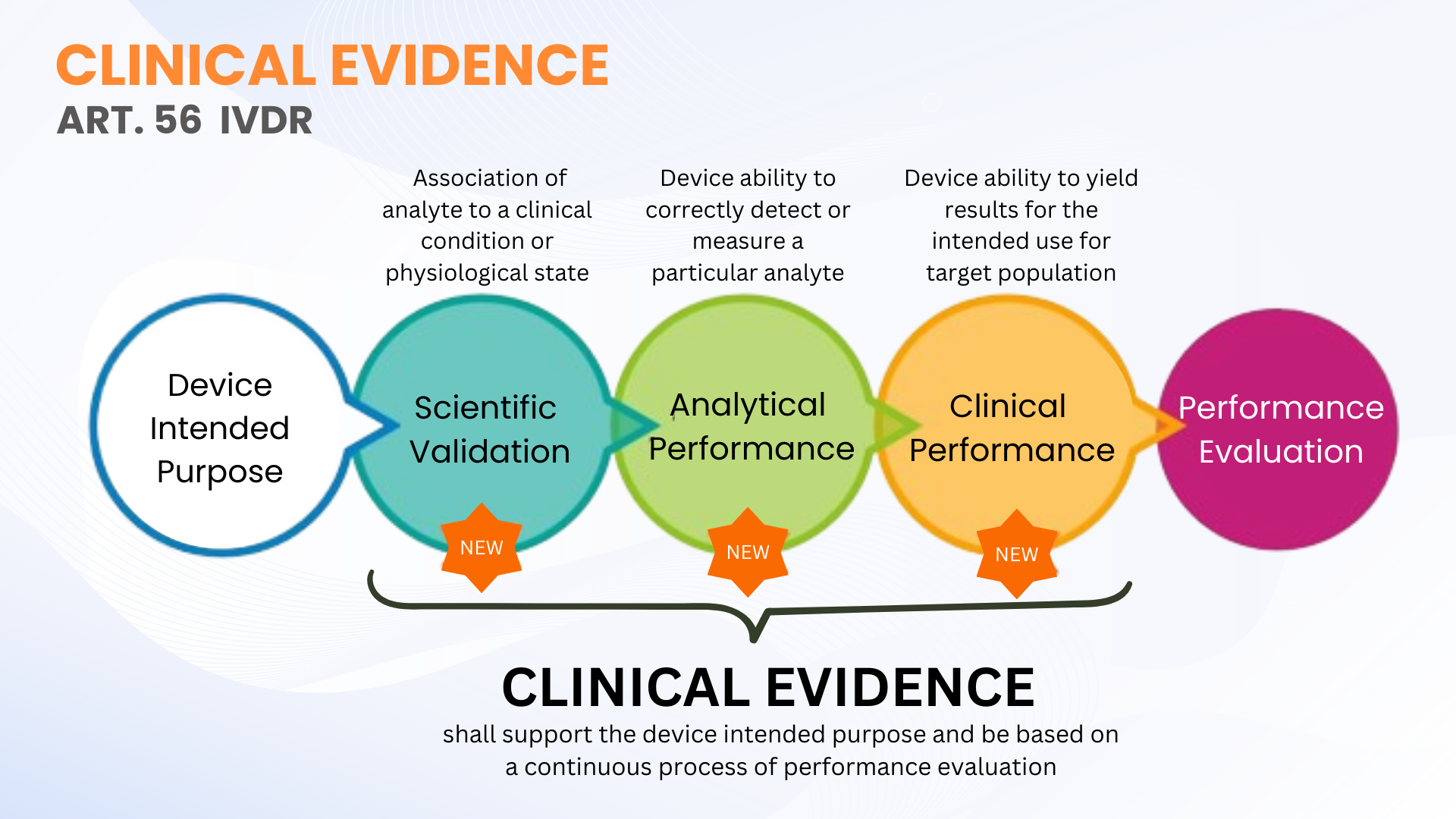
IVDR 没有定义特定IVD 设备要测试的临床样本的具体数量(一些高风险IVD 参数除外,这些参数在通用规范中定义)。这种灵活性使 IVD 制造商能够计划和进行适合其设备的 PE 测试。然而,制造商应计划扩大其临床性能研究方法,以根据设备的风险分类来支持临床证据。风险等级越高,就越有可能需要临床表现研究来支持临床证据。
PE 文件和验证的重要性
IVD 制造商必须在进行 PE 测试之前,根据 IVDR 附件 XIII A 部分第 1.1 节定义并提交 PEP,因为不这样做可能会损害测试数据的完整性。如果没有证据表明他们的 PE 测试遵循原始PEP(和预定义的验收标准),NB 审核人员可能会得出结论,PEP 是事后修改的,以证实测试结果。 NB 审核员可以轻松验证和跟踪 PEP 何时发布、如何遵循以及何时完成 PE 测试。如果这个时间表不明确,通常会引发一些问题,例如:PE流程是否没有明确定义?为什么没有按照 IVDR 的要求进行记录?为什么没有包括 PE 数据分析的验收标准和方法?因此,为了使您的设计和开发以及CE标记过程更加高效,IVD公司应严格遵循IVDR中明确定义的PE文档流程和要求。
许多IVD 制造商都在努力实施结构化但灵活高效的 PE 流程,该流程不会限制设计和开发活动的创新。对于许多公司来说,在开始 PE 活动之前制定一个描述其 PE 活动的QMS 程序或制定一个明确定义的 PE 计划的简单要求仍然是一个重大挑战。这通常与QMS流程、培训以及对IVDR和解释指南(例如MDCG 2022-2)中定义的PE要求的理解不充分有关。
PE 报告必须详尽
PER 中 PE 结果、结论和基本原理的充分记录对于制造商 TDA 的成功至关重要,因为 NB 审核员不得代表制造商解释任何数据。如果文件和理由没有明确表达,这可能会导致 NB 评审员做出最坏情况的估计,并需要进行额外的临床性能测试或 PMPF 研究。检查充分性的一个好方法是问自己是否会根据 PEP 和 PER 中展示的 PE 数据和客观证据来发布 IVD 设备。如果您没有内部能力,合格的临床评估人员可以支持您的 PER 的最终编写,以确保其满足 NB 审核员的要求。
传统设备可能需要新的临床证据来支持修改后的预期用途
旧IVDs 的制造商在 IVDR 下修改和/或扩展预期目的,可能需要重新定义其整体 PE 活动、科学有效性、分析性能和临床性能测试,以证明临床证据。在某些情况下,如果在设计和开发设备时尚未建立 PE 流程,则可能需要追溯创建 PEP。如果是这样,制造商应在其PE流程中清楚地描述这种追溯方法,并根据IVDR要求和最新标准重写PEP和PER。通常,对 PEP 和 PER 进行实质性更新需要对所进行的调整和变更提供详细的理由和风险评估。如果不这样做,制造商可能会面临 NB 审查人员的重大问题。
如果 PE 信息质量优良并且有明确的记录和验证,许多这些情况都可以轻松避免。利用收集到的上市后监测 (PMS) 数据可以进一步支持临床证据,例如特定技术或标记物。 PMS数据还可以验证该技术或设备是否完善并且已受到制造商的良好控制。这进一步降低了设备的整体风险,从而有助于您的 NB 审查取得成功。
如果您在建立 PE 流程、将您的 QMS 更新至 IVDR 或编写 PE 文档方面需要支持,Pure Global 可以提供帮助。我们的 SMART 软件工具简化了科学数据的收集和评估,以及在 EU 市场上选择类似或等效设备。了解更多关于我们的绩效评估支持的信息。



无论您身在何处,
都欢迎联系我们。
无论您是在寻找更多信息,还是已经准备与我们合作,我们都会在监管流程的每一步提供指导。
联系我们