
IVDR の臨床証拠: IVD メーカーにとっての「適切なレベル」とは何ですか?
IVDR に準拠するには、臨床証拠の品質に対する厳密なアプローチが必要です。この記事では、オリバー アイケンバーグ博士が、EU IVDR コンプライアンスの適切な臨床証拠を確保するためのパフォーマンス評価プロセスと文書の重要性について説明します。
臨床証拠は、体外診断機器規制 (EU IVDR 2017/746) に基づく新しい要件ではありません。ただし、IVDD とは対照的に、より明確に定義されています。 IVDR の第 56 条と附属書 XIII では、臨床証拠を、科学的妥当性、分析性能、臨床性能を含むIVDs のパフォーマンス評価 (PE)の継続的なプロセスとして定義しています。より簡単に言うと、臨床証拠は、IVD が安全であり、意図された目的に従って機能することを実証するためのパフォーマンス評価のデータと所見で構成されます。
IVDR の意図された目的の基準が拡大され、安全性全般に対してより厳格なアプローチが導入されたことを考慮すると、多くの IVDR メーカーは、特に IVDR の移行期限が近づくにつれ、適切な臨床証拠を確実に提示するためにどこまで取り組む必要があるのか疑問に思っています。
しかし、多くの IVD メーカーは、慎重な PE プロセスの必要性や、明確に評価および分析された PE データと適切なレベルの情報を認証機関 (NB) に提供する利点を依然として過小評価しています。臨床証拠は安全で効果的な機器の全体的な決定基準であるため、徹底した PE 文書化、プロセス、リスクベースの正確なデータ分析が NB レビューに合格するための鍵となります。
IVDRにおける業績評価とは何ですか?
パフォーマンス評価 (PE) は、臨床証拠をサポートするメカニズムです。これは、意図された目的に対するデバイスの科学的妥当性、分析性能、および臨床性能を実証するために、データが評価および分析される、適切に構造化された透明性のある反復的かつ継続的なプロセスである必要があります。これはメーカーの QMS の一部である必要があり、市販後を含むライフサイクル全体を通じて発生します。
他の多くのプロセスと同様、PE は計画から始まります。パフォーマンス評価計画 (PEP) では、データ収集方法、タイムライン、承認基準、および担当者の責任を定義する必要があります。 PE テストからの PE データ収集は、PEP に従って実行されます。利用可能な科学データまたは PE データは、定義されたデータ分析に基づいて収集および評価 (評価) され、その結論はパフォーマンス評価レポート (PER) に通知されます。市販後の活動には、最先端技術(規格など)の継続的な監視と評価、および市販後性能フォローアップ (PMPF) 活動が含まれ、これらは該当する場合、製造元の PMPF レポートおよび定期安全更新レポート (PSUR) に文書化されます。リスク管理はこれらすべての活動の中核であり、各ステップに含める必要があります。
MDCG 2022-2 は、PE プロセスを簡略化したグラフで示しています。
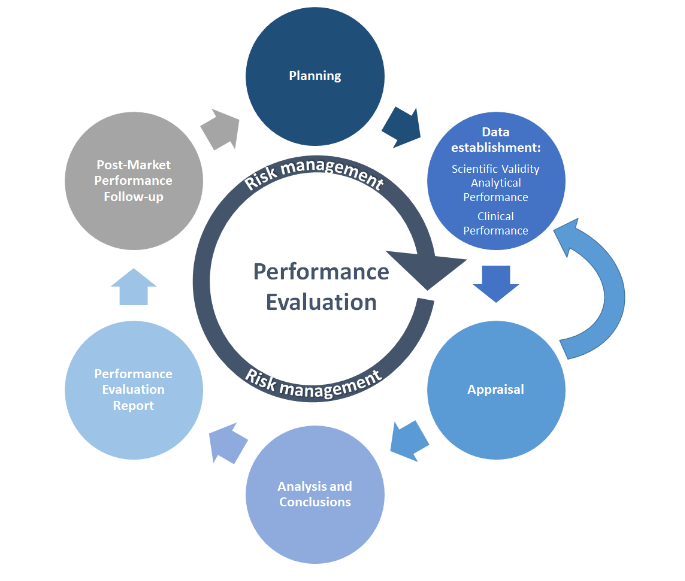
ソース: MDCG 2022-2
前述したように、IVDs の臨床証拠は、科学的妥当性、分析パフォーマンス、臨床パフォーマンスからの PE データの収集を通じて確立できます。証拠は、デバイスの安全性と、製造業者の意図どおりに使用した場合に意図された臨床上の利点が達成されるかどうかについて、認定された技術文書評価 (公認機関による TDA) を可能にするレベルまで収集および文書化される必要があります。これは必ずしも臨床成績研究を実施しなければならないという意味ではありません。場合によっては、資格のある評価者による科学的妥当性の評価と、適切な数の臨床サンプルを使用した分析的 PE テストで十分な場合があります。
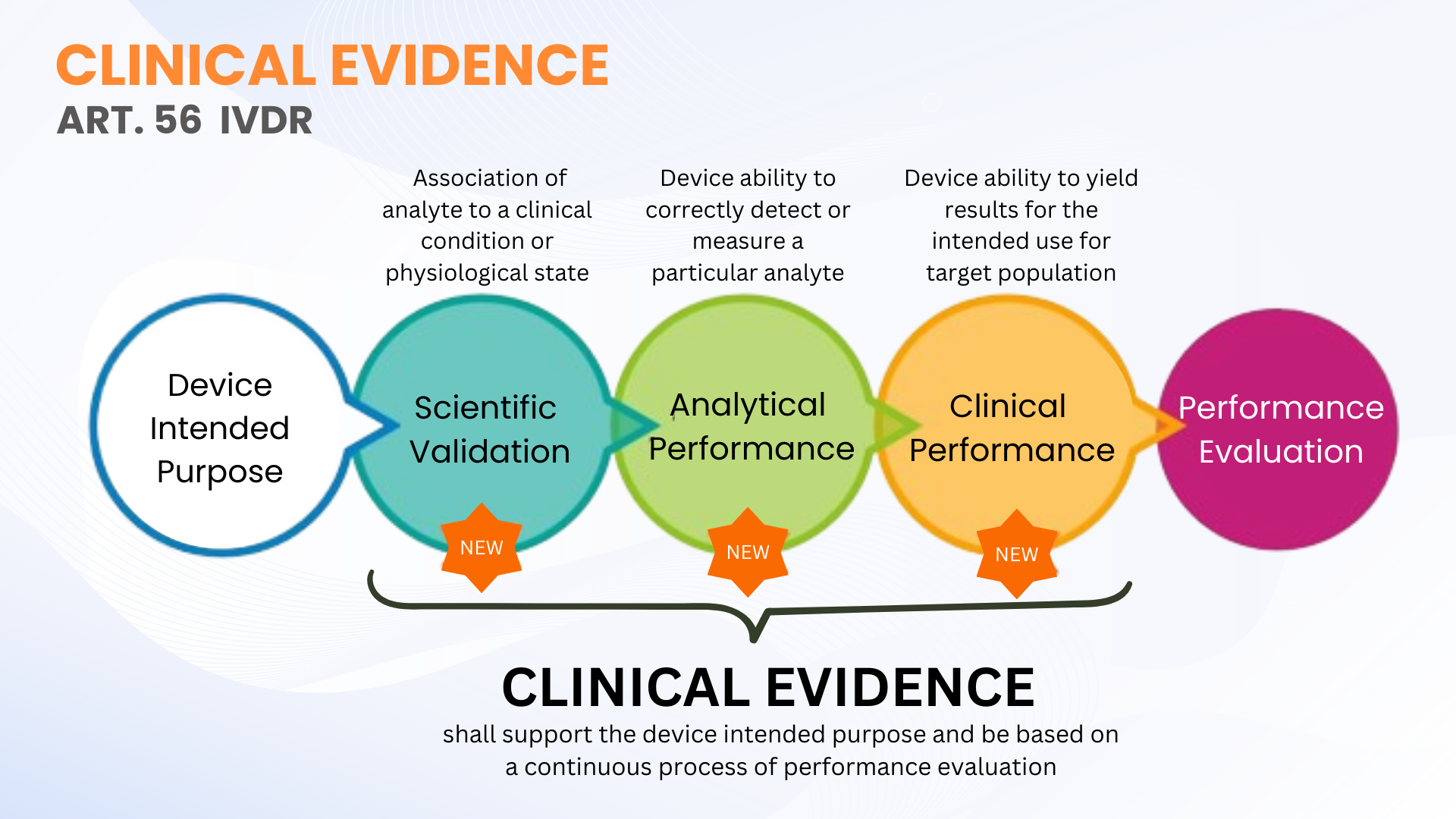
IVDR では、特定の IVD デバイスについて検査される臨床サンプルの具体的な数は定義されていません (共通仕様で定義されている一部の高リスク IVD パラメーターを除く)。この柔軟性により、IVD メーカーは自社のデバイスに適した PE テストを計画および実施できます。ただし、メーカーは、機器のリスク分類に従って臨床証拠を裏付けるために、臨床性能研究のアプローチを拡大することを計画する必要があります。リスククラスが高いほど、臨床的証拠を裏付けるために臨床成績研究が必要になる可能性が高くなります。
PEの文書化と検証の重要性
IVD メーカーは、PE テストを実施する前に、IVDR、付録 XIII、パート A、セクション 1.1 に従って PEP を定義し、提出する必要があります。これを怠ると、テスト データの整合性が損なわれる可能性があるためです。 PE テストが元の PEP (および事前に定義された合格基準) に従っていたという証拠がなければ、NB の審査担当者は、テスト結果を実証するために PEP が事後修正されたと結論付ける可能性があります。 NB 監査人は、PEP がいつリリースされたか、どのように遵守されたか、PE テストがいつ行われたかを簡単に検証および追跡できます。このタイムラインが明確でない場合、通常、次のような疑問が生じます。PE プロセスは明確に定義されていないのですか? IVDR で必須として文書化されていないのはなぜですか? PE データ分析の受け入れ基準と方法が含まれていないのはなぜですか?したがって、設計と開発、および CE マーキング プロセスをより効率的にするには、IVD 企業は、IVDR で明確に定義された PE 文書化プロセスと要件に厳密に従う必要があります。
多くの IVD 製造業者は、設計と開発活動によるイノベーションを制限しない、構造化された機敏で効率的な PE プロセスの実装に苦労しています。 PE 活動を説明する QMS 手順を作成するか、PE 活動を開始する前に明確に定義された PE 計画を作成するという単純な要件は、多くの企業にとって依然として大きな課題です。これは多くの場合、不適切な QMS プロセス、トレーニング、IVDR および解釈ガイドライン (MDCG 2022-2 など) で定義された PE 要件の理解に関連しています。
PE レポートは徹底する必要があります
NB 監査人はメーカーに代わっていかなるデータも解釈することは許可されていないため、PER に PE の結果、結論、理論的根拠を適切に文書化することは、メーカーの TDA の成功にとって重要です。文書と理論的根拠が明確に表現されていない場合、NB 審査員による最悪の場合の推定が行われ、追加の臨床性能試験や PMPF 研究の実施が必要になる可能性があります。適切性をチェックする良い方法は、PEP と PER で実証された PE データと客観的な証拠に基づいて、IVD デバイスを解放するかどうか自問することです。社内に能力がない場合は、資格のある臨床評価者が PER の最終執筆をサポートし、NB 監査人の要件を満たしていることを確認します。
レガシーデバイスには、改訂された意図された目的をサポートするための新しい臨床証拠が必要になる可能性があります
IVDR に基づいて改訂または拡張された意図された目的に取り組んでいる従来の IVDs のメーカーは、臨床的証拠を証明するために全体的な PE 活動、科学的妥当性、分析パフォーマンス、および臨床パフォーマンス テストを再定義する必要がある可能性があります。デバイスの設計および開発時に PE プロセスがまだ確立されていなかった場合、PEP を遡及的に作成する必要がある場合があります。その場合、メーカーはこの遡及的アプローチを PE プロセスに明確に記述し、IVDR の要件と最先端の規格に従って PEP と PER を書き直す必要があります。通常、PEP および PER に対する大幅な更新には、実施された適応と変更の詳細な正当化とリスク評価が必要です。これが行われない場合、メーカーは NB 審査員から重大な質問に直面する可能性があります。
PE 情報が高品質であり、明確に文書化され検証されていれば、これらのシナリオの多くは簡単に回避できます。収集した市販後調査 (PMS) データを活用することで、特定の技術やマーカーなどの臨床証拠をさらに裏付けることができます。 PMS データは、テクノロジーやデバイスが十分に確立されており、製造元によって適切に管理されていることを検証することもできます。これにより、デバイスの全体的なリスクがさらに軽減され、NB レビューの成功に貢献します。
PE プロセスの確立、QMS の IVDR への更新、または PE ドキュメントの作成に関するサポートが必要な場合は、Pure Global がお手伝いします。当社の SMART ソフトウェア ツールは、EU 市場での類似または同等のデバイスの選択だけでなく、科学データの収集と評価を効率化します。当社の性能評価支援について詳しくはこちらをご覧ください。



どこにいても、
ご相談ください。
詳しい情報が必要な段階でも、協業を始める段階でも、規制対応の各ステップをサポートします。
お問い合わせ