
Klinische Evidenz unter IVDR: Was ist ein „angemessenes Niveau“ für IVD-Hersteller?
Die Konformität mit dem IVDR erfordert einen strengen Ansatz für die Qualität klinischer Beweise. In diesem Artikel erläutert Dr. Oliver Eikenberg die Bedeutung von Leistungsbewertungsprozessen und Dokumentation, um angemessene klinische Beweise für die Einhaltung von EU und IVDR sicherzustellen.
Klinische Beweise sind keine neue Anforderung gemäß der Verordnung über In-vitro-Diagnostika (EU IVDR 2017/746); Im Gegensatz zum IVDD ist es jedoch klarer definiert. Artikel 56 und Anhang XIII des IVDR definieren klinische Beweise als einen kontinuierlichen Prozess der Leistungsbewertung (PE) für IVDs, der wissenschaftliche Validität, analytische Leistung und klinische Leistung umfasst. Vereinfacht ausgedrückt umfassen klinische Beweise die Daten und Erkenntnisse aus der Leistungsbewertung, um zu zeigen, dass Ihr IVD sicher ist und seinen beabsichtigten Zweck erfüllt.
Angesichts der erweiterten IVDR-Kriterien für den beabsichtigten Zweck und der Einführung eines strengeren Sicherheitsansatzes im Allgemeinen fragen sich viele IVDR-Hersteller, wie weit sie gehen müssen, um sicherzustellen, dass sie ausreichende klinische Beweise vorlegen, insbesondere da die Übergangsfristen für IVDR näher rücken.
Viele IVD-Hersteller unterschätzen jedoch immer noch die Notwendigkeit eines sorgfältigen PE-Prozesses sowie die Vorteile der Bereitstellung klar bewerteter und analysierter PE-Daten und eines angemessenen Informationsniveaus für benannte Stellen (NBs). Da klinische Beweise das allgemeine Entscheidungskriterium für ein sicheres und wirksames Gerät sind, sind eine gründliche PE-Dokumentation, ein Prozess und eine risikobasierte, genaue Datenanalyse der Schlüssel zum Bestehen einer NB-Prüfung.
Was ist die Leistungsbewertung unter IVDR?
Die Leistungsbewertung (PE) ist der Mechanismus, der Ihre klinischen Beweise stützt. Es sollte ein gut strukturierter, transparenter, iterativer und kontinuierlicher Prozess sein, durch den Daten bewertet und analysiert werden, um die wissenschaftliche Gültigkeit, die analytische Leistung und die klinische Leistung des Geräts für seinen beabsichtigten Zweck nachzuweisen. Es muss Teil des QMS des Herstellers sein und erfolgt während des gesamten Lebenszyklus, einschließlich Post-Market.
Wie viele andere Prozesse beginnt PE mit der Planung. Der Leistungsbewertungsplan (PEP) muss die Datenerfassungsmethoden, Zeitpläne, Akzeptanzkriterien und Personalverantwortlichkeiten definieren. Die PE-Datenerfassung aus PE-Tests wird dann gemäß PEP durchgeführt. Verfügbare wissenschaftliche oder PE-Daten werden gesammelt und auf der Grundlage der definierten Datenanalyse bewertet (bewertet), deren Schlussfolgerungen in den Leistungsbewertungsbericht (PER) einfließen. Post-Market-Aktivitäten umfassen die kontinuierliche Überwachung und Bewertung des Stands der Technik (z. B. Standards) sowie Aktivitäten zur Nachverfolgung der Leistung nach dem Inverkehrbringen (PMPF), die gegebenenfalls im PMPF-Bericht und im Periodic Safety Update Report (PSUR) des Herstellers dokumentiert werden. Das Risikomanagement ist der Kern all dieser Aktivitäten und sollte in jeden Schritt einbezogen werden.
MDCG 2022-2 veranschaulicht den PE-Prozess in einer vereinfachten Grafik:
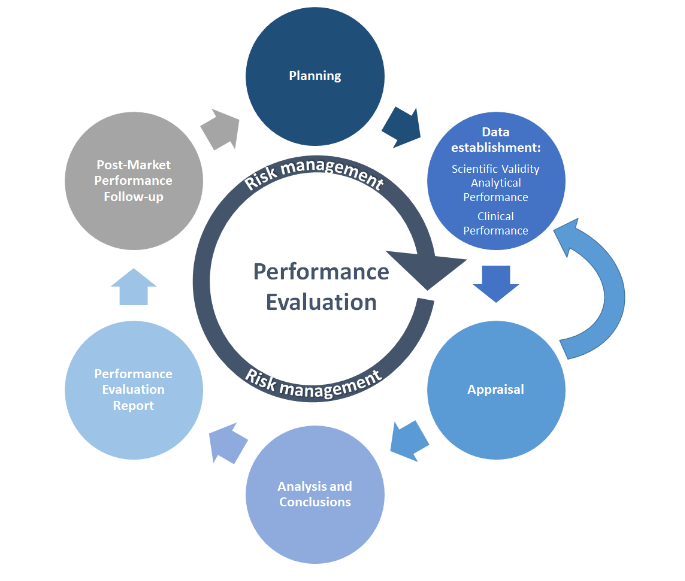
Quelle: MDCG 2022-2
Wie bereits erwähnt, können klinische Beweise für IVDs durch die Sammlung von PE-Daten aus wissenschaftlicher Validität, analytischer Leistung und klinischer Leistung erbracht werden. Die Nachweise sollten auf einem Niveau gesammelt und dokumentiert werden, das eine qualifizierte Bewertung der technischen Dokumentation (TDA durch eine benannte Stelle) der Sicherheit des Geräts und der Frage, ob es bei bestimmungsgemäßer Verwendung des Herstellers den beabsichtigten klinischen Nutzen erzielt, ermöglicht. Dies bedeutet nicht unbedingt, dass klinische Leistungsstudien durchgeführt werden müssen. Manchmal reichen eine wissenschaftliche Validitätsbewertung durch qualifizierte Gutachter und analytische PE-Tests mit einer ausreichenden Anzahl klinischer Proben aus.
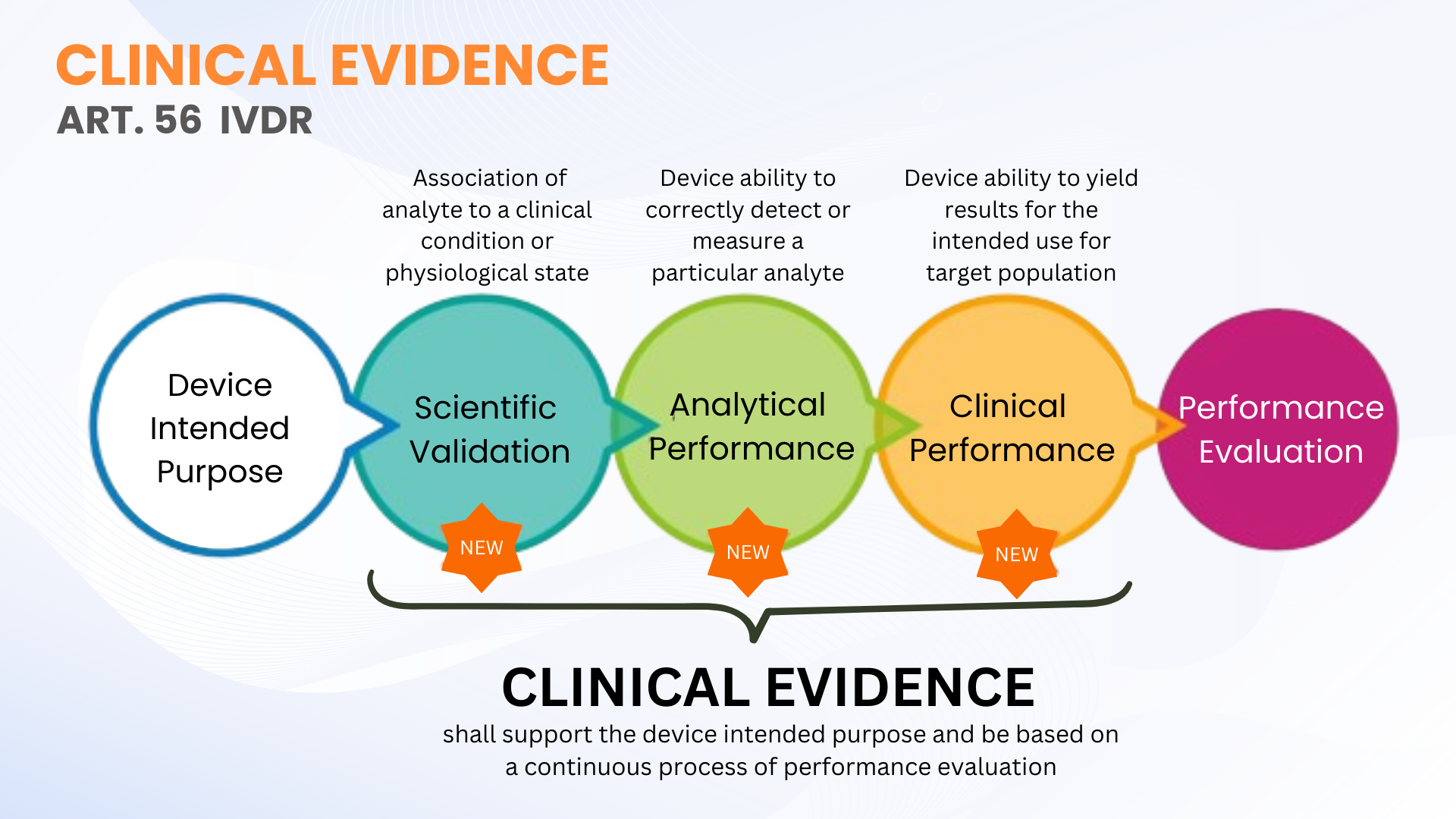
Das IVDR definiert keine spezifische Anzahl klinischer Proben, die für bestimmte IVD-Geräte getestet werden sollen (mit Ausnahme einiger IVD-Parameter mit hohem Risiko, wo diese in den Common Specifications definiert sind). Diese Flexibilität ermöglicht es IVD-Herstellern, die für ihr Gerät geeigneten PE-Tests zu planen und durchzuführen. Hersteller sollten jedoch planen, ihren Ansatz für klinische Leistungsstudien zu skalieren, um die klinischen Beweise entsprechend der Risikoklassifizierung des Geräts zu untermauern. Je höher die Risikoklasse, desto wahrscheinlicher ist es, dass klinische Leistungsstudien zur Untermauerung der klinischen Evidenz erforderlich sind.
Die Bedeutung der PE-Dokumentation und -Überprüfung
IVD-Hersteller müssen ein PEP gemäß IVDR, Anhang XIII, Teil A, Abschnitt 1.1 definieren und einreichen, bevor sie PE-Tests durchführen, da ein Versäumnis die Integrität ihrer Testdaten gefährden könnte. Ohne Beweise dafür, dass ihre PE-Tests dem ursprünglichen PEP (und den vordefinierten Akzeptanzkriterien) folgten, könnten NB-Prüfer zu dem Schluss kommen, dass der PEP nachträglich überarbeitet wurde, um die Testergebnisse zu untermauern. NB-Auditoren können leicht überprüfen und nachverfolgen, wann PEPs freigegeben wurden, wie sie befolgt wurden und wann PE-Tests durchgeführt wurden. Wenn dieser Zeitplan nicht klar ist, wirft dies normalerweise Fragen auf, wie zum Beispiel: Ist der PE-Prozess nicht klar definiert? Warum ist es nicht wie erforderlich unter IVDR dokumentiert? Warum sind Akzeptanzkriterien und Methoden für die PE-Datenanalyse nicht enthalten? Um Ihr Design und Ihre Entwicklung sowie den CE-Markierungsprozess effizienter zu gestalten, sollten IVD-Unternehmen daher die klar definierten PE-Dokumentationsprozesse und -Anforderungen im IVDR strikt befolgen.
Viele IVD-Hersteller haben Schwierigkeiten, einen strukturierten, aber agilen und effizienten PE-Prozess zu implementieren, der die Innovation aus Design- und Entwicklungsaktivitäten nicht einschränkt. Die einfache Anforderung, über ein QMS-Verfahren zur Beschreibung ihrer PE-Aktivitäten zu verfügen oder vor Beginn der PE-Aktivitäten einen klar definierten PE-Plan zu haben, stellt für viele Unternehmen immer noch eine große Herausforderung dar. Dies hängt oft mit unzureichenden QMS-Prozessen, Schulungen und unzureichendem Verständnis der PE-Anforderungen zusammen, die im IVDR und in Interpretationsrichtlinien wie MDCG 2022-2 definiert sind.
Die PE-Berichterstattung muss gründlich sein
Eine angemessene Dokumentation der PE-Ergebnisse, Schlussfolgerungen und Begründungen im PER ist entscheidend für den Erfolg der TDA des Herstellers, da NB-Auditoren nicht berechtigt sind, Daten im Namen des Herstellers zu interpretieren. Wenn die Dokumentation und die Begründungen nicht klar zum Ausdruck gebracht werden, könnte dies zu einer Worst-Case-Schätzung durch NB-Prüfer führen, die die Durchführung zusätzlicher klinischer Leistungstests oder PMPF-Studien erforderlich macht. Eine gute Methode zur Überprüfung der Angemessenheit besteht darin, sich zu fragen, ob Sie Ihr IVD-Gerät basierend auf den PE-Daten und objektiven Beweisen, die in Ihrem PEP und PER nachgewiesen wurden, freigeben würden. Wenn Sie nicht über die entsprechenden Kapazitäten verfügen, können qualifizierte klinische Gutachter Sie bei der endgültigen Erstellung Ihres PER unterstützen, um sicherzustellen, dass es den Anforderungen des NB-Auditors entspricht.
Ältere Geräte benötigen möglicherweise neue klinische Beweise, um einen überarbeiteten Verwendungszweck zu unterstützen
Hersteller von älteren IVDs, die mit einem überarbeiteten und/oder erweiterten Verwendungszweck im Rahmen des IVDR arbeiten, müssen möglicherweise ihre gesamten PE-Aktivitäten, wissenschaftliche Validität, analytische Leistung und klinische Leistungstests neu definieren, um die klinische Evidenz nachzuweisen. In einigen Fällen muss das PEP möglicherweise nachträglich erstellt werden, wenn der PE-Prozess zum Zeitpunkt der Konzeption und Entwicklung des Geräts noch nicht etabliert war. Wenn ja, sollten Hersteller diesen retrospektiven Ansatz in ihrem PE-Prozess klar beschreiben und das PEP und PER gemäß den IVDR-Anforderungen und den neuesten Standards neu schreiben. Typischerweise erfordern umfangreiche Aktualisierungen des PEP und PER detaillierte Begründungen und Risikobewertungen für die durchgeführten Anpassungen und Änderungen. Geschieht dies nicht, könnten die Hersteller mit erheblichen Fragen seitens der NB-Rezensenten konfrontiert werden.
Viele dieser Szenarien können leicht vermieden werden, wenn PE-Informationen von höchster Qualität sind und klar dokumentiert und überprüft werden. Die Nutzung der gesammelten Daten zur Post-Market-Überwachung (PMS) kann die klinischen Beweise – etwa für eine bestimmte Technologie oder einen bestimmten Marker – weiter unterstützen. PMS-Daten können auch bestätigen, dass die Technologie oder das Gerät gut etabliert ist und vom Hersteller gut kontrolliert wurde. Dadurch wird das Gesamtrisiko für das Gerät weiter reduziert, was zum Erfolg Ihrer NB-Bewertung beiträgt.
Wenn Sie Unterstützung beim Aufbau von PE-Prozessen, bei der Aktualisierung Ihres QMS auf IVDR oder beim Verfassen der PE-Dokumentation benötigen, kann Pure Global helfen. Unsere SMART-Softwaretools optimieren die Sammlung und Auswertung wissenschaftlicher Daten sowie die Auswahl ähnlicher oder gleichwertiger Geräte auf dem EU-Markt. Erfahren Sie mehr über unsere Unterstützung bei der Leistungsbewertung.



Sprechen wir,
wo immer Sie sind.
Ob Sie weitere Informationen suchen oder bereit zur Zusammenarbeit sind: Wir begleiten Sie durch jeden Schritt des regulatorischen Prozesses.
Kontakt