
Evidencia clínica bajo IVDR: ¿qué es un “nivel adecuado” para los fabricantes de IVD?
La conformidad con el IVDR requiere un enfoque riguroso de la calidad de la evidencia clínica. En este artículo, el Dr. Oliver Eikenberg explica la importancia de los procesos y la documentación de evaluación del desempeño para garantizar evidencia clínica adecuada para el cumplimiento de EU IVDR.
La evidencia clínica no es un requisito nuevo según el Reglamento de dispositivos de diagnóstico in vitro (EU IVDR 2017/746); sin embargo, está más claramente definido en contraste con IVDD. El artículo 56 y el Anexo XIII del IVDR definen la evidencia clínica como un proceso continuo de evaluación de desempeño (PE) para IVDs que incluye validez científica, desempeño analítico y desempeño clínico. En términos más simples, la evidencia clínica comprende los datos y los hallazgos de la evaluación de desempeño para demostrar que su IVD es seguro y funciona de acuerdo con el propósito previsto.
Teniendo en cuenta que los criterios de IVDR ampliaron el propósito previsto e introdujeron un enfoque más riguroso para la seguridad en general, muchos fabricantes de IVDR se preguntan hasta dónde deben llegar para garantizar que presentan evidencia clínica adecuada, especialmente a medida que se acercan los plazos de transición de IVDR.
Sin embargo, muchos fabricantes de IVD todavía subestiman la necesidad de un proceso de PE cuidadoso, así como los beneficios de proporcionar datos de PE claramente evaluados y analizados y un nivel adecuado de información a los Organismos Notificados (ON). Debido a que la evidencia clínica es el criterio de decisión general para un dispositivo seguro y eficaz, la documentación exhaustiva de PE, el proceso y el análisis de datos precisos basados en el riesgo son las claves para aprobar una revisión de NB.
¿Qué es la evaluación del desempeño según IVDR?
La evaluación del desempeño (PE) es el mecanismo que respalda su evidencia clínica. Debe ser un proceso bien estructurado, transparente, iterativo y continuo mediante el cual se evalúen y analicen los datos para demostrar la validez científica, el desempeño analítico y el desempeño clínico del dispositivo para el propósito previsto. Debe ser parte del QMS del fabricante y ocurre durante todo el ciclo de vida, incluida la poscomercialización.
Como muchos otros procesos, la educación física comienza con la planificación. El Plan de Evaluación del Desempeño (PEP) debe definir los métodos de recolección de datos, cronogramas, criterios de aceptación y responsabilidades del personal. Luego, la recopilación de datos de PE a partir de las pruebas de PE se realiza de acuerdo con PEP. Los datos científicos o de educación física disponibles se recopilan y evalúan (valoran) en función del análisis de datos definido, cuyas conclusiones informarán el Informe de evaluación del desempeño (PER). Las actividades posteriores a la comercialización cubren el seguimiento y la evaluación continuos del estado de la técnica (por ejemplo, estándares), así como las actividades de seguimiento del desempeño posterior a la comercialización (PMPF), documentadas en el Informe PMPF del fabricante y el Informe periódico de actualización de seguridad (PSUR), según corresponda. La gestión de riesgos es el núcleo de todas estas actividades y debe incluirse en cada paso.
MDCG 2022-2 ilustra el proceso de PE en un gráfico simplificado:
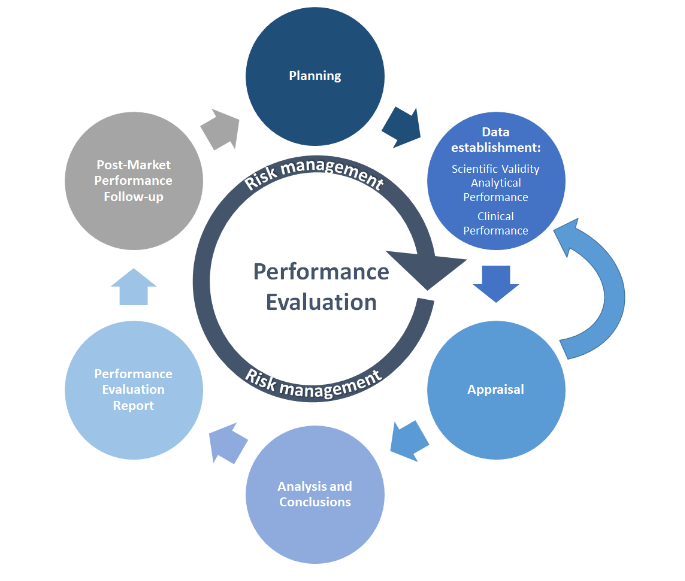
Fuente: MDCG 2022-2
Como se indicó, la evidencia clínica de IVDs se puede establecer mediante la recopilación de datos de PE a partir de la validez científica, el desempeño analítico y el desempeño clínico. La evidencia debe recopilarse y documentarse a un nivel que permita una evaluación de la documentación técnica calificada (TDA por organismo notificado) de la seguridad del dispositivo y si logra los beneficios clínicos previstos cuando se utiliza según lo previsto por el fabricante. Esto no significa necesariamente que se deban realizar estudios de rendimiento clínico. En ocasiones, es suficiente la evaluación de la validez científica a través de evaluadores calificados y pruebas analíticas de PE con un número adecuado de muestras clínicas.
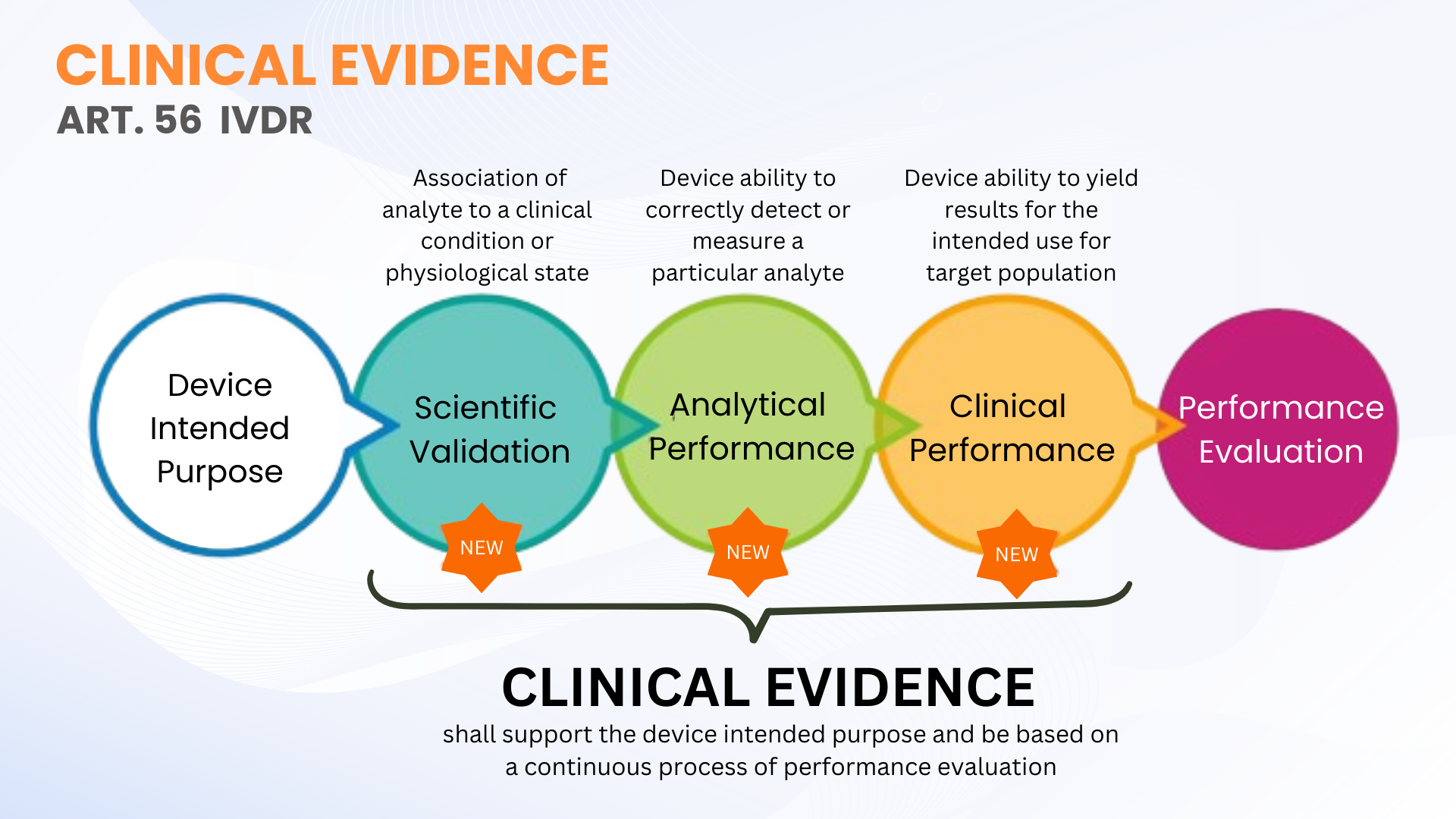
IVDR no define números específicos de muestras clínicas que se analizarán para ciertos dispositivos IVD (excepto algunos parámetros IVD de alto riesgo, donde se definen en Especificaciones comunes). Esta flexibilidad permite a los fabricantes de IVD planificar y realizar las pruebas de PE adecuadas para su dispositivo. Sin embargo, los fabricantes deben planificar la ampliación de su enfoque de estudio de rendimiento clínico para respaldar la evidencia clínica de acuerdo con la clasificación de riesgo del dispositivo. Cuanto mayor sea la clase de riesgo, más probable será que se necesiten estudios de rendimiento clínico para respaldar la evidencia clínica.
La importancia de la documentación y verificación de PE
Los fabricantes de IVD deben definir y presentar un PEP de acuerdo con IVDR, Anexo XIII, parte A, sección 1.1 antes de realizar pruebas de PE, ya que no hacerlo podría comprometer la integridad de los datos de sus pruebas. Sin evidencia de que sus pruebas de PE siguieron el PEP original (y los criterios de aceptación predefinidos), los revisores de NB pueden concluir que el PEP fue revisado después del hecho para fundamentar los resultados de la prueba. Los auditores de NB pueden verificar y rastrear fácilmente cuándo se publicaron las PEP, cómo se siguieron y cuándo se realizaron las pruebas de PE. Si este cronograma no está claro, generalmente surgen preguntas como: ¿No está claramente definido el proceso de EP? ¿Por qué no está documentado según lo exige IVDR? ¿Por qué no se incluyen los criterios y métodos de aceptación para el análisis de datos de PE? Por lo tanto, para que su diseño y desarrollo y el proceso de marcado CE sean más eficientes, las empresas de IVD deben seguir estrictamente los procesos y requisitos de documentación de PE bien definidos en IVDR.
Muchos fabricantes de IVD luchan por implementar un proceso de PE estructurado pero ágil y eficiente que no limite la innovación de las actividades de Diseño y Desarrollo. El simple requisito de tener un procedimiento QMS que describa sus actividades de PE o tener un plan de PE claramente definido antes de iniciar las actividades de PE sigue siendo un desafío importante para muchas empresas. Esto a menudo está relacionado con procesos, capacitación y comprensión inadecuados de QMS de los requisitos de PE definidos en IVDR y en las pautas de interpretación, como MDCG 2022-2.
Los informes de PE deben ser exhaustivos
La documentación adecuada de los resultados, conclusiones y fundamentos de PE en el PER es fundamental para el éxito del TDA del fabricante, ya que los auditores NB no pueden interpretar ningún dato en nombre del fabricante. Si la documentación y los fundamentos no se expresan claramente, esto podría resultar en una estimación del peor de los casos por parte de los revisores de NB con la necesidad de realizar pruebas de rendimiento clínico adicionales o estudios PMPF. Un buen método para comprobar la idoneidad es preguntarse si liberaría su dispositivo IVD basándose en los datos PE y la evidencia objetiva demostrada en su PEP y PER. Si no tiene las capacidades internas, los evaluadores clínicos calificados pueden respaldar la redacción final de su PER para garantizar que cumpla con los requisitos del auditor de NB.
Los dispositivos heredados podrían necesitar nueva evidencia clínica para respaldar un propósito previsto revisado
Los fabricantes de IVDs heredados que trabajan con un propósito previsto revisado o ampliado según el IVDR podrían necesitar redefinir sus actividades generales de PE, su validez científica, su desempeño analítico y sus pruebas de desempeño clínico para demostrar la evidencia clínica. En algunos casos, es posible que sea necesario crear el PEP de forma retrospectiva, si el proceso de PE aún no se había establecido en el momento en que se diseñó y desarrolló el dispositivo. Si es así, los fabricantes deben describir claramente este enfoque retrospectivo en su proceso de PE y reescribir PEP y PER de acuerdo con los requisitos de IVDR y los estándares más modernos. Normalmente, las actualizaciones sustanciales de PEP y PER requieren justificaciones detalladas y evaluaciones de riesgos para las adaptaciones y cambios realizados. Si esto no se hace, los fabricantes pueden enfrentar preguntas importantes por parte de los revisores de NB.
Muchos de estos escenarios pueden evitarse fácilmente si la información de PE es de calidad superior y está claramente documentada y verificada. La utilización de los datos de vigilancia posterior a la comercialización (PMS) recopilados puede respaldar aún más la evidencia clínica, como una tecnología o un marcador específicos. Los datos PMS también pueden verificar que la tecnología o el dispositivo está bien establecido y que ha sido bien controlado por el fabricante. Esto reduce aún más el riesgo general para el dispositivo, lo que contribuye al éxito de su revisión NB.
Si necesita ayuda para establecer procesos de PE, actualizar su QMS a IVDR o redactar la documentación de PE, Pure Global puede ayudar. Nuestras herramientas de software SMART agilizan la recopilación y evaluación de datos científicos, así como la selección de dispositivos similares o equivalentes en el mercado EU. Conozca más sobre nuestro apoyo a la evaluación del desempeño.



Hablemos,
esté donde esté.
Ya sea que busque más información o esté listo para asociarse con nosotros, le guiaremos en cada paso del proceso regulatorio.
Contáctenos