借助 AI 驱动的注册文档编制和本地代理服务,加速市场准入,按固定年费收费。首个器械每年 2,000 美元起(不含政府费用)。包含文档提交、本地代理、翻译、变更、经销商授权及上市后支持。
Malaysia's MDA and China's NMPA have launched Pilot Phase 2 of the Malaysia-China Joint Evaluation Pilot Programme, open from 1 July to 30 September 2026. The programme enables simultaneous evaluation of eligible medical devices under the GHWP CERP framework to reduce duplicative review and support faster access to both markets.
From launching AI-driven regulatory solutions to expanding our global footprint, it was a year of meaningful milestones and shared moments.
新加坡健康科学局 (HSA) 现已成为全球公认的参考机构,为制造商提供进入亚太主要市场的快速通道。本文解释HSA批准如何简化泰国、澳大利亚和香港的注册,以及其世界卫生组织严格监管机构地位如何促进高风险IVDs的准入。利用 HSA 可以削减成本、缩短时间并加速国际扩张。
销售无线医疗器械的制造商必须考虑通信机构的法规以及医疗器械的法规。在本文中,Michael Cassidy 解释了制造商在将无线模块集成到其设备时需要考虑的全球市场准入要求。
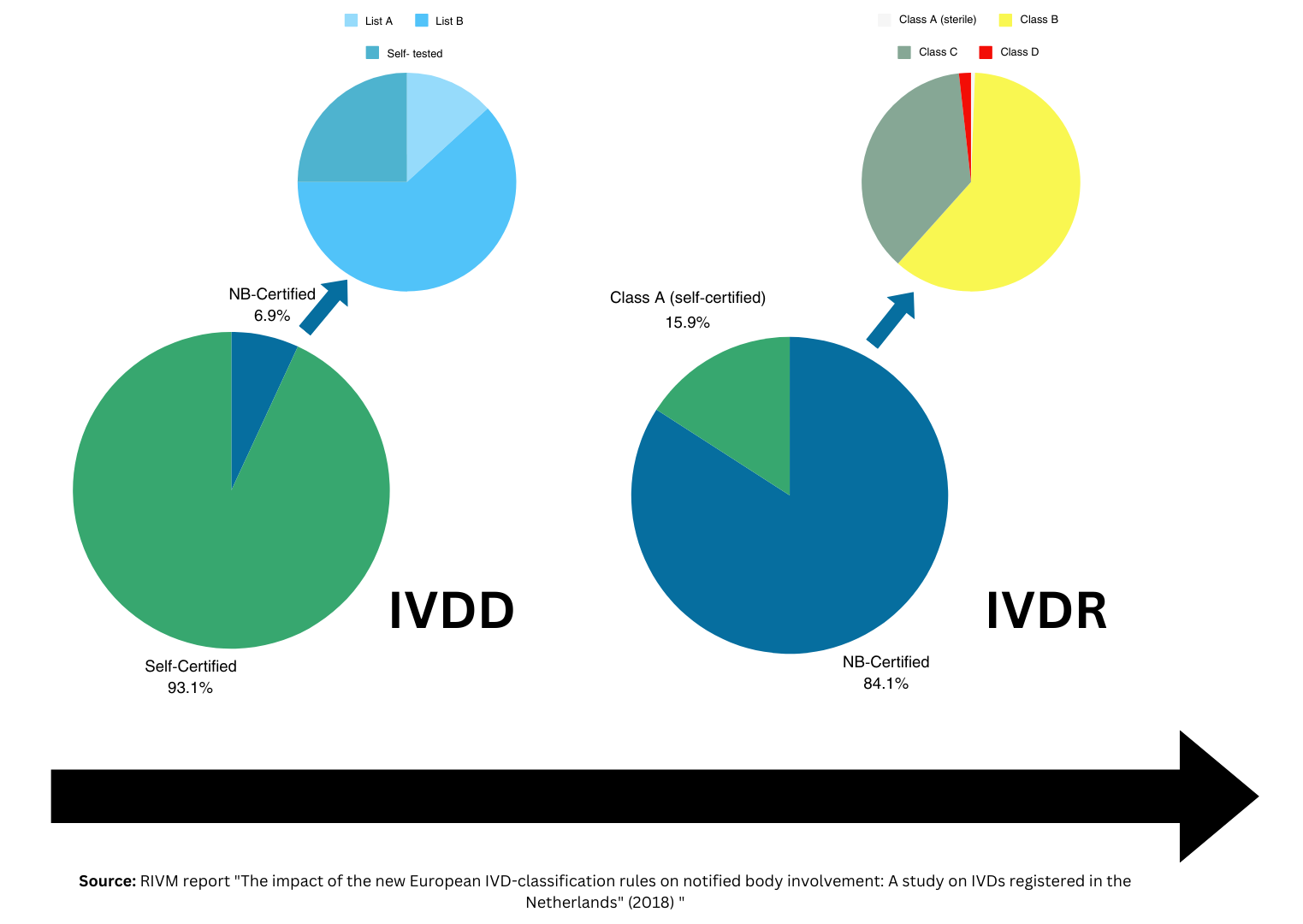
根据IVDR,欧洲CE 标记需要IVD 制造商比以往更多的时间和资源。然而,随着越来越多的国家采用 EU 的医疗器械方法和 IVD 监管,IVD 公司将发现,一旦 IVDR CE 标志就位,他们将能够很好地进入 EU 以外的其他市场。在本文中,Oliver Eikenberg 博士讨论了利用 IVDR CE 标记简化全球市场准入的潜力。
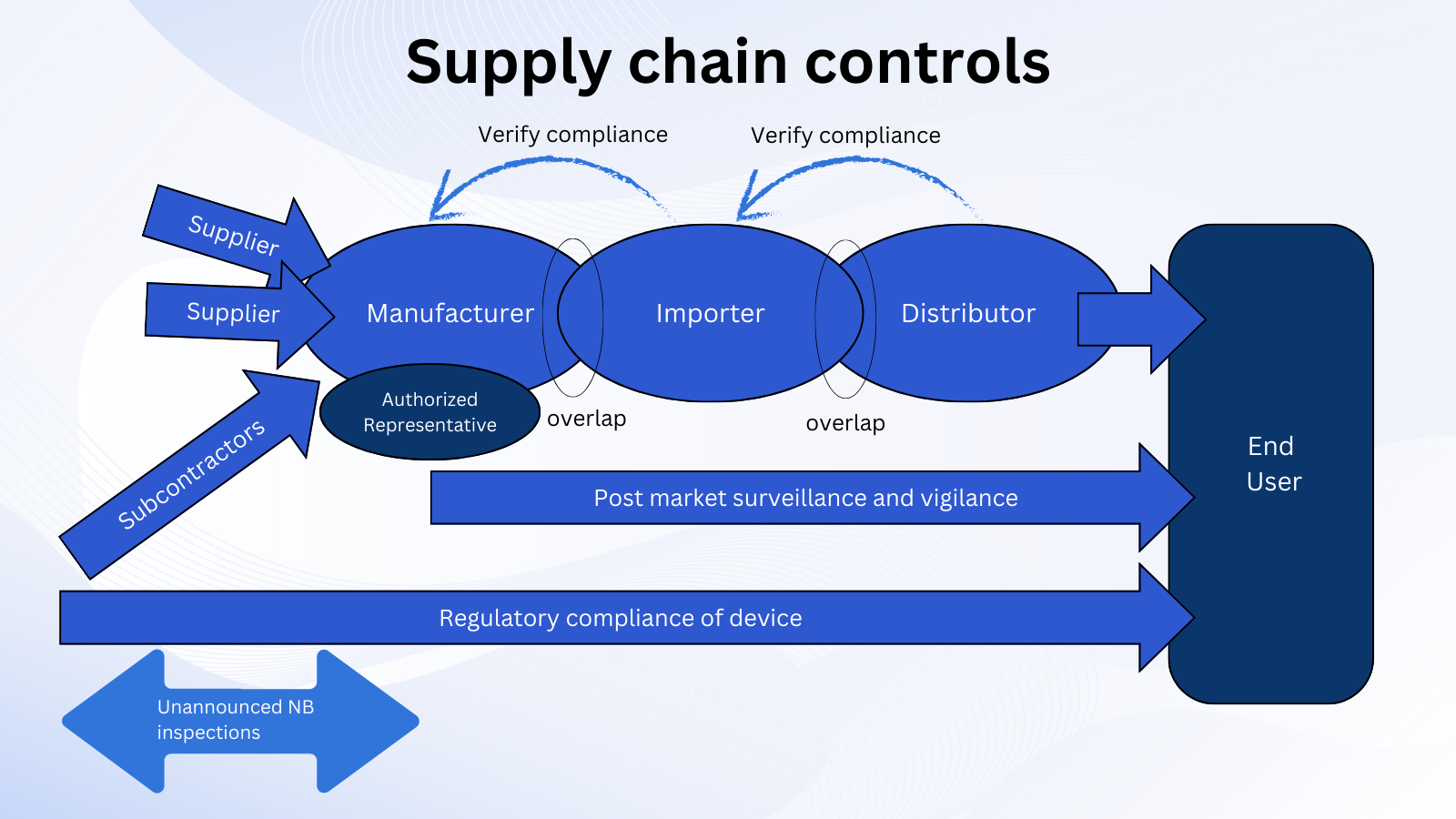
供应链控制不足是 IVDR 下公告机构技术文件评估不合格的主要原因。在本文中,Oliver Eikenberg 博士讨论了制造商应如何评估和更新其供应链协议,以确保经济运营商的角色和义务按照 IVDR 要求进行明确定义。
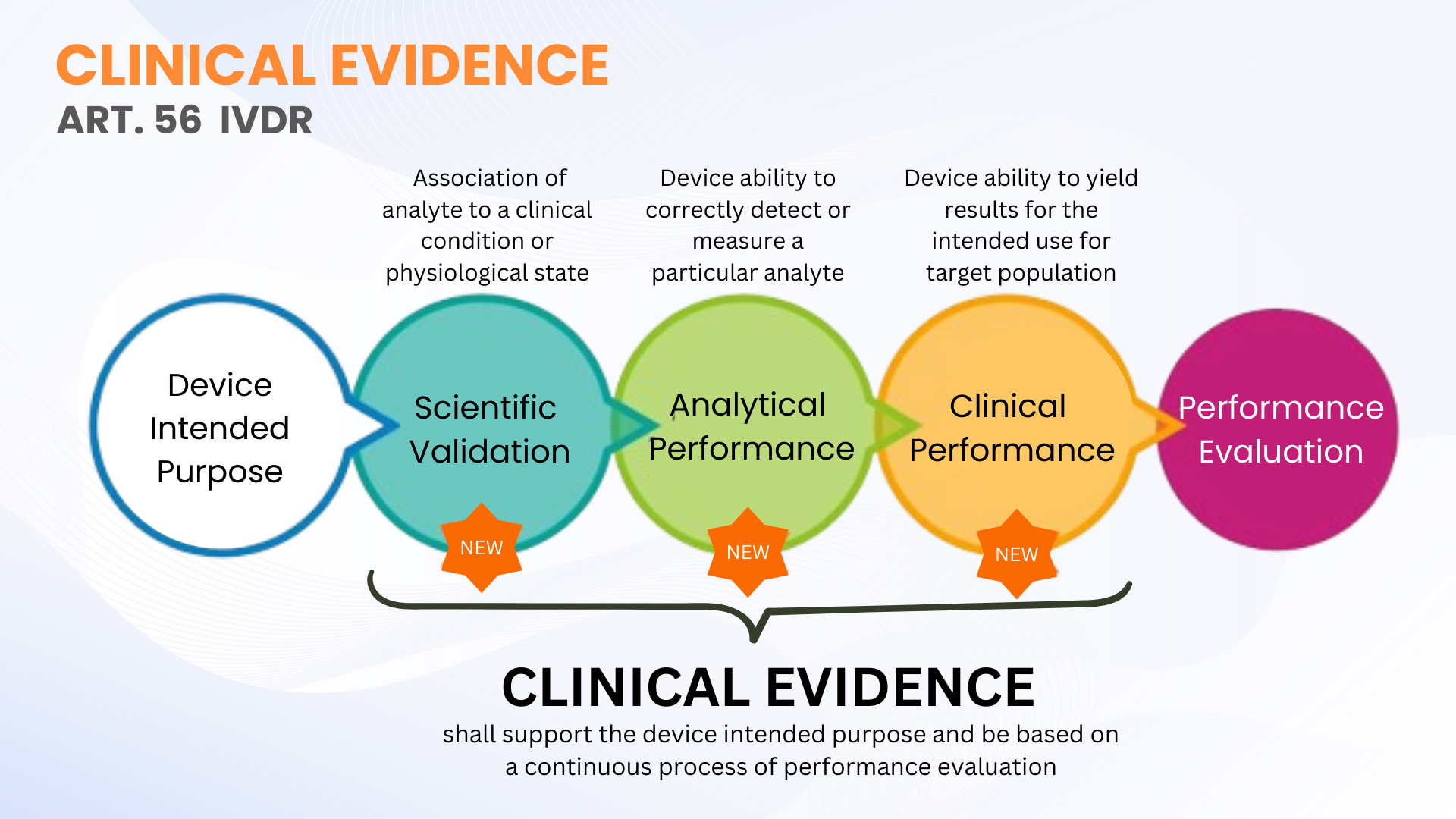
遵守 IVDR 要求对临床证据质量采取严格的方法。在本文中,Oliver Eikenberg 博士解释了绩效评估流程和文档的重要性,以确保 EU IVDR 合规性有足够的临床证据。
IVDR 过渡包括IVD 制造商必须遵循的结构化流程才能与公告机构合作。许多IVD 制造商是第一次与公告机构互动,必须了解确保成功合作伙伴关系所需的条件。在本文中,Oliver Eikenberg 博士阐明了制造商必须准备什么以及何时聘请公告机构进行 IVDR CE 标记。
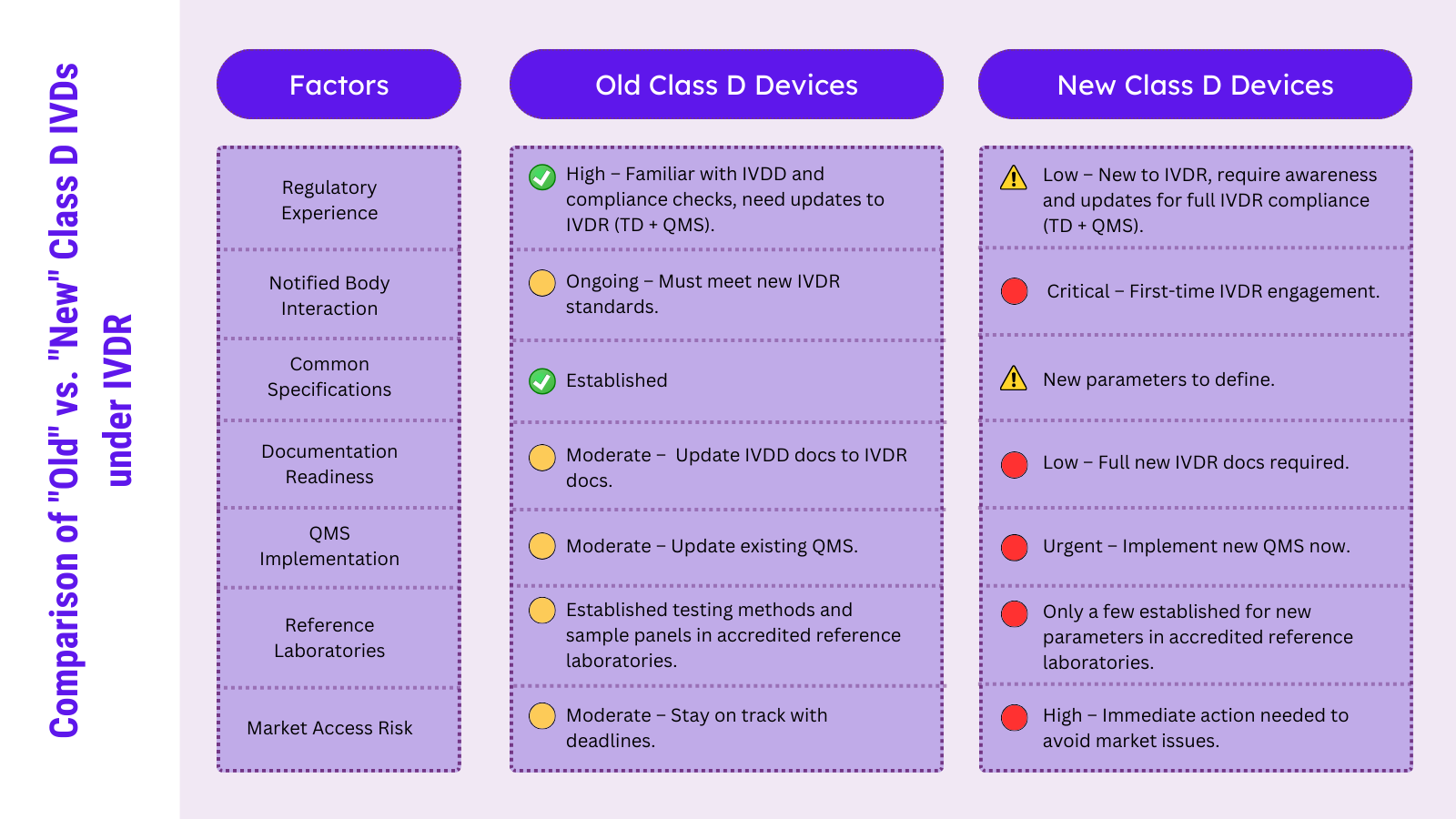
D 类 IVD 制造商面临着快速临近的 IVDR 过渡期限,尽管有些制造商更熟悉监管流程。在本文中,Oliver Eikenberg 博士讨论了“新”和“旧”D 类之间的区别,以及它们在过渡过程中需要考虑的监管逻辑。
IVD 的预期用途决定了IVDR 下的分类、临床证据要求和临床应用。设备公司将需要更强大的预期目的来满足 IVDR 标准。在本文中,Oliver Eikenberg 博士解释了预期目的在 IVDR 合规性中的作用。
许多IVD 和传统IVD 设备制造商甚至在过渡截止日期之前就有义务遵守IVDR 的某些方面。在本文中,Oliver Eikenberg 讨论了IVD 公司如何在过渡期间保持EU 合规性。
无论您是在寻找更多信息,还是已经准备与我们合作,我们都会在监管流程的每一步提供指导。