
IVDR에 따른 신규 및 기존 클래스 D 장치에 대한 규제 요구 사항 및 과제
클래스 D IVD 제조업체는 IVDR 전환 기한이 빠르게 다가오고 있지만 일부 제조업체는 규제 프로세스에 더 익숙합니다. 이 기사에서 Oliver Eikenberg 박사는 "신규" 클래스와 "기존" 클래스 D의 차이점과 전환 과정에서 고려해야 할 규제 물류에 대해 논의합니다.
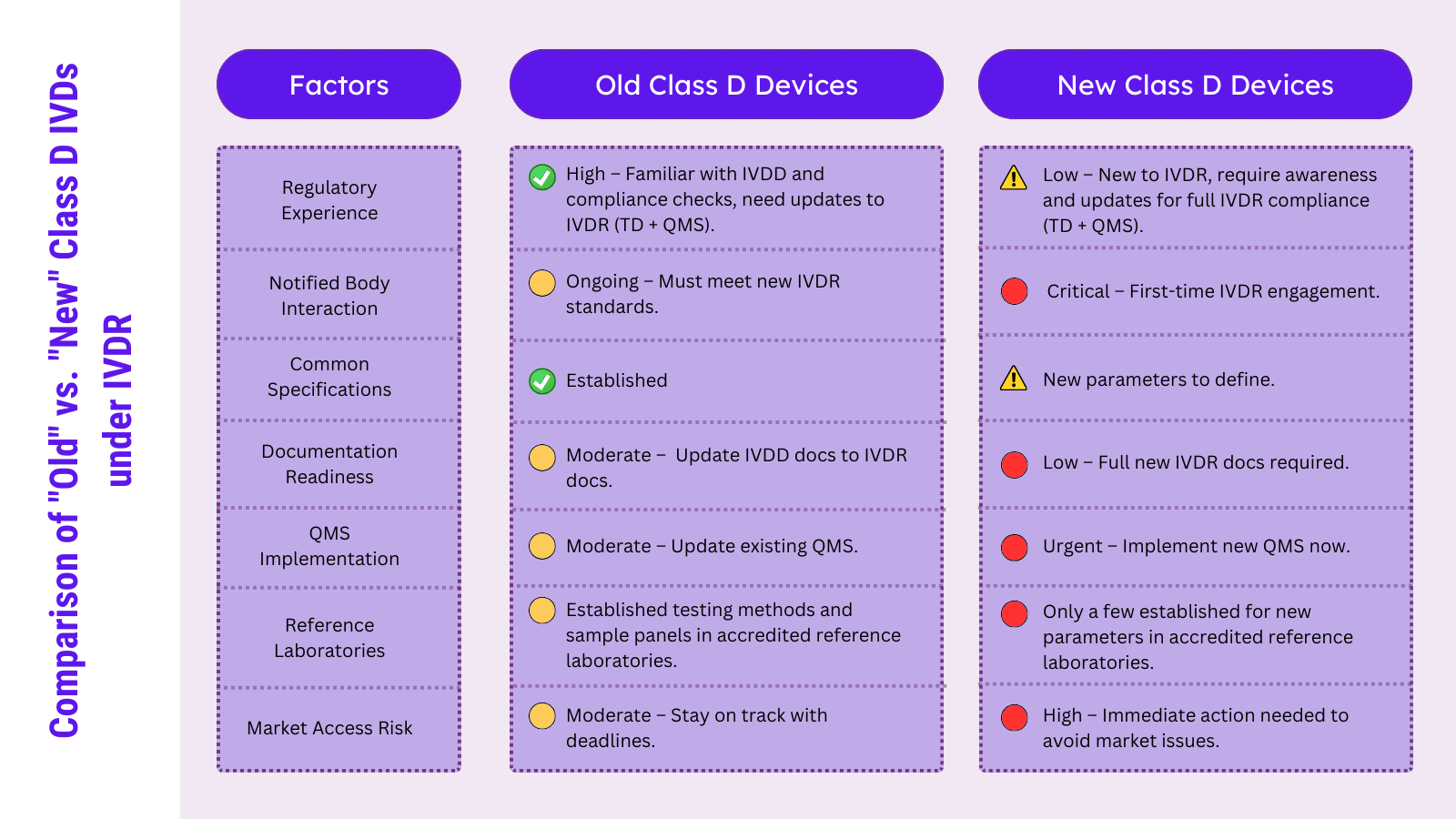
IVDR에 따라 클래스 D 장치는 이제 두 가지 광범위한 범주로 분류됩니다. 이전에 IVDD의 목록 A 및 B에 포함된 장치(예: HIV, 간염, ABO, 혈액형 분류, 톡소플라스마, 거대 세포 바이러스, 클라미디아, 풍진, PSA, 혈당 자가 검사)와 IVDR에 새로 분류된 장치(예: SARS-CoV-2, 말라리아, 뎅기열, 치쿤구니야, 지카 바이러스, 트레포네마 팔리듐, 트리파노소마 크루지). 두 그룹(“기존” 및 “신규”) 모두 임시 전환 요구 사항을 준수하고 인증 기관 운영을 고려해야 합니다.
첫 번째 그룹은 IVDD(인증 기관이 발행한 확장 EC 인증서)에 따라 기존 규제 경험과 가능한 레거시 상태의 혜택을 받는 반면, 두 번째 그룹은 일반적으로 인증 기관의 피드백과 동일한 수준의 규제 경험이 없습니다. 클래스 D로 새로 분류된 장치 제조업체는 많은 새로운 요구 사항을 준수해야 한다고 느낄 수 있으며 이는 기존 클래스 D 장치 제조업체보다 더 까다로울 수 있습니다.
"기존" 클래스 D IVD 장치
클래스 D 장치의 첫 번째 범주는 인증기관의 완전한 통제를 받고 있거나 곧 통제하게 될 것입니다. IVDD의 인증 기관이 레거시 장치로 확장한 현재 EC 인증서는 곧 종료됩니다. IVDR을 통해 구현된 새로운 프로세스와 공식적으로 설립된 새로운 EU 참조 실험실이 있더라도 이러한 클래스 D 매개변수에 대한 전체 프로세스와 경험은 IVDD과 유사해야 합니다. 대부분의 클래스 D 매개변수에 대해 공통 사양이 설정되어 있으므로 EU 참조 실험실은 필요한 성능 테스트에 대한 장기적인 경험을 보유하고 있습니다. 전반적으로 기존 클래스 D 장치 제조업체의 주요 활동은 QMS를 구현 및 업데이트하고 기술 문서를 IVDR에 의해 도입된 의무에 맞게 갱신하는 것입니다.
"새로운" 클래스 D IVD 장치
IVD 장치의 두 번째 그룹은 IVDD에서 처리되지 않은 새로운 분석 물질과 클레임으로 구성됩니다. 이러한 장치 중 상당 수는 IVDD에 따라 "일반" IVDs에 포함될 수 있으며 IVDR의 과도기적 의무가 충족된다면 여전히 레거시 장치로 판매될 수 있습니다. 이러한 IVD 제조업체는 이미 인증기관과 연락하고 있을 수 있습니다. 또한 처음으로 기술 문서 및 QMS에 대한 독립적인 감사를 받아야 합니다. 이러한 독립적인 인증 기관 피드백은 많은 질문을 제기할 수 있으며 제조업체와 인증 기관 간의 IVDR 의무에 대한 적절하고 객관적인 문서 및 해석에 대한 서로 다른 이해를 식별하는 경우가 많습니다.
또한 공통 사양과 EU 참조 실험실은 몇 가지 매개변수에 대해서만 확립되었습니다(예: Treponema pallidum, Trypanosoma cruzi, SARS-CoV-2의 경우 (EU) 2022/1107). 다음 달에는 추가 매개변수가 뒤따를 수 있지만, 이 새로운 프로세스에 관련된 모든 당사자의 작업과 질문은 기존 마커보다 시간이 더 많이 소요될 것으로 예상됩니다. 이러한 이유로 이 그룹의 IVD 제조업체는 이러한 변화에 대비하고 EU 시장을 유지하기 위해 지금 조치를 취해야 합니다.
클래스 D IVD 제조업체가 인증 기관과 조기에 연락해야 하는 이유
2024년 7월 9일, MDR 및 IVDR에 대한 새로운 개정안 (EU) 2024/1860이 게시되었으며, 무엇보다도 특정 IVDs(레거시 장치)에 대한 새로운 확장된 과도기 조항이 도입되었습니다. 이는 2017년(IVDR이 게시된 날짜) 이후 전환 일정이 연장된 세 번째 수정안입니다. 이 개정안에 따라 클래스 D 장치에는 다음 요구 사항과 함께 2027년 12월 31일의 새로운 전환 기한이 적용됩니다.
제조업체는 늦어도 2025년 5월 26일까지 IVDR 준수 QMS을 마련해야 합니다.
제조업체는 늦어도 2025년 5월 26일까지 인증기관에 IVDR 신청서를 제출해야 합니다.
제조업체는 늦어도 2025년 9월 26일까지 인증 기관과 공식 서면 계약을 체결해야 합니다.
이전 수정안에서는 IVDR 제출과 관련된 작업량을 고려하지 않았기 때문에 일정과 의무는 2024/1860년 세 번째 수정안(EU)에 구체적으로 구현되었습니다. 많은 수가 마지막 순간(마감일 몇 달 전)에 제출되었습니다. 인증 기관은 이를 관리할 수 없었고 잠재적인 IVD 부족 위험이 증가했습니다. 따라서 유럽연합 집행위원회는 의료 서비스의 원활한 기능에 대한 잠재적 영향과 EU 시장에 미치는 영향에 대해 논의했습니다. 이전 개정으로 인해 개정된 규정(EU) 2024/1860은 제조업체와 인증 기관에 활동을 수행할 수 있는 추가 시간을 제공했지만 인증 기관 역량을 신청할 수 있는 마감일과 제한 사항이 정의되어 있었습니다.
인증 기관의 입장에 서십시오. 인증 기관은 클래스 D 제조업체를 지원하고 IVDR에 따라 복잡한 적합성 프로세스를 관리하기 위해 적절한 일정에 걸쳐 예상되는 작업량을 설명해야 합니다. 따라서 인증 기관은 이러한 기한을 준수하는 제조업체만이 IVDR 전환 일정이 길어지는 혜택을 누릴 수 있다고 전달했습니다. 지연은 제출물이 검토 대기열 끝에 추가되어 적합성 평가가 지연될 위험에 직면하게 됨을 의미합니다.
클래스 D IVD 제조업체에 대한 이러한 기한은 IVDR 준수 QMS을 마련하고 IVDR 신청서를 인증 기관에 제출하고 (2025년 9월) 인증 기관과 공식 서면 계약에 서명해야 하는 의무로 2025년 5월부터 시작됩니다. 현재 사용 가능한 인증 기관의 역량을 최대한 활용하려면 이러한 일정을 적절하게 준비하고 충족하는 것이 현명합니다.
우리는 IVDR으로의 전환 프로젝트가 얼마나 어렵고 시간이 많이 걸릴 수 있는지 알고 있습니다. 특히 필요한 기술 문서 수준과 QMS을 처음 접하는 경우라면 더욱 그렇습니다. 철저한 준비와 효율성이 중요하지만 모든 제조업체는 IVD 장치를 EU 시장에 계속 출시하려면 지금이 조치를 취해야 할 때라는 점을 인식해야 합니다.
Pure Global은 장치 클래스 확인부터 기술 문서 제출, QMS 구현 및 인증 기관과의 상호 작용까지 맞춤형 지원을 제공합니다. 저희 IVDR 컨설팅 서비스에 대해 자세히 알아보세요.



어디에 계시든,
상담해 드립니다.
더 많은 정보가 필요하든 협업을 시작할 준비가 되었든, 규제 절차의 모든 단계를 안내합니다.
문의하기