
IVDR の新旧クラス D デバイスの規制要件と課題
クラス D IVD の製造業者は、IVDR の移行期限が迫っていますが、中には規制プロセスに精通しているメーカーもあります。この記事では、オリバー アイケンバーグ博士が、「新しい」クラス D と「古い」クラス D の違い、および移行プロセス中に考慮する必要がある規制上のロジスティクスについて説明します。
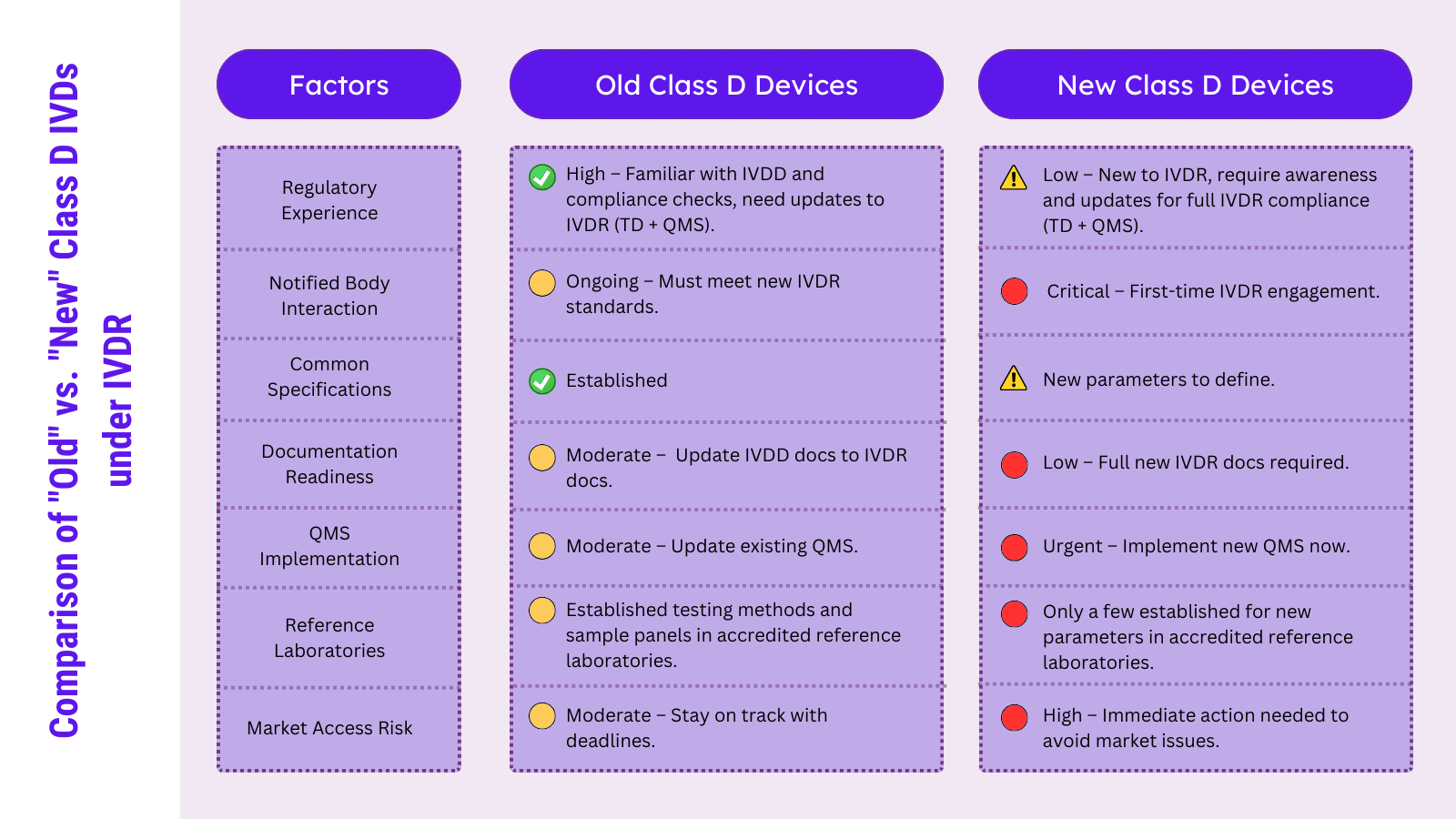
IVDR の下で、クラス D デバイスは 2 つの大きなカテゴリに分類されます。1 つは以前に IVDD のリスト A および B でカバーされていたもの (例: HIV、肝炎、ABO、血液型検査、トキソプラズマ、サイトメガロウイルス、クラミジア、風疹、PSA、血糖自己検査)、および新たに IVDR に分類されたもの (例: SARS-CoV-2、マラリア、デング熱、チクングニア熱、ジカウイルス、梅毒トレポネーマ、クルーズトリパノソーマ)。両方のグループ (「古い」グループと「新しい」グループ) は、暫定的な移行要件を遵守し、公認機関の運営を検討する必要があります。
最初のグループは既存の規制経験と IVDD (認証機関によって発行された拡張 EC 証明書) に基づくレガシー ステータスの可能性から恩恵を受けていますが、2 番目のグループは通常、同レベルの規制経験と認証機関からのフィードバックを持っていません。新たにクラス D に分類されたデバイスのメーカーは、多くの新しい要件に準拠する必要があると感じる可能性があり、これは確立されたクラス D デバイスのメーカーよりも困難になる可能性があります。
「古い」クラス D IVD デバイス
クラス D デバイスの最初のカテゴリは、現在、または間もなく完全に認証機関の管理下に置かれる予定です。 IVDD の認証機関によってレガシー デバイスとして拡張された現在の EC 証明書は間もなく終了します。正式に設立された IVDR および新しい EU 参照研究所を通じて新しいプロセスが実装されたとしても、これらのクラス D パラメーターの全体的なプロセスとエクスペリエンスは IVDD と比較して類似しているはずです。これらのクラス D パラメータのほとんどについて共通の仕様が確立されているため、EU 参照研究所は、必要な性能試験に関する長期の経験を持っています。全体として、古いクラス D デバイスのメーカーの主な活動は、QMS を導入・更新し、技術文書を IVDR によって導入された義務に適合させることです。
「新しい」クラス D IVD デバイス
IVD デバイスの 2 番目のグループは、IVDD では対処されていない新しい分析物とクレームで構成されています。 IVDD によれば、これらのデバイスのかなりの数が「一般」IVDs の対象となる可能性があり、IVDR の移行義務が満たされていれば、引き続きレガシー デバイスとして販売される可能性があります。これらの IVD メーカーは、すでに認証機関と連絡を取っている可能性があります。また、技術文書と QMS について初めて独立した監査を受ける必要があります。この独立した認証機関からのフィードバックは多くの疑問を引き起こす可能性があり、メーカーと認証機関の間で適切かつ客観的な文書化と IVDR の義務の解釈についての異なる理解が判明することがよくあります。
さらに、共通仕様と EU 参照研究所は、いくつかのパラメーターについてのみ確立されています (例: 梅毒トレポネーマ、クルーズトリパノソーマ、SARS-CoV-2 の (EU) 2022/1107)。来月には追加のパラメータが追加される可能性がありますが、この新しいプロセスに関与するすべての関係者からの作業と質問は、確立されたマーカーの場合よりも時間がかかることが予想されます。このため、このグループの IVD メーカーは、この変化に備えて EU 市場に留まるために今すぐ行動する必要があります。
クラス D IVD の製造業者が早期に認証機関に連絡する必要がある理由
2024 年 7 月 9 日、MDR および IVDR に対する新たな修正 (EU) 2024/1860 が公開され、特に特定の IVDs (レガシー デバイス) に対する新しい拡張された移行規定が導入されました。これは、2017 年 (IVDR が公開された日) 以来、移行スケジュールが延長された 3 回目の修正です。この修正により、クラス D デバイスには次の要件とともに、2027 年 12 月 31 日という新たな移行期限が設定されます。
メーカーは 2025 年 5 月 26 日までに、IVDR に準拠した QMS を導入する必要があります。
製造業者は 2025 年 5 月 26 日までに認証機関に IVDR 申請書を提出しなければなりません
製造業者は 2025 年 9 月 26 日までに、認証機関と正式な書面による契約を締結する必要があります。
以前の修正では IVDR の提出に関連する作業負荷が考慮されていなかったため、タイムラインと義務はこの修正第 3 修正 (EU) 2024/1860 で具体的に実装されています。多くは土壇場(締め切りの数か月前)に提出されました。認証機関はそれらを管理できず、潜在的な IVD 不足のリスクが増大しました。したがって、欧州委員会は、医療サービスの円滑な機能に対する潜在的な影響と、EU 市場への影響について議論しました。以前の修正により、修正された規制 (EU) 2024/1860 では、製造業者と認証機関に活動を行うための追加の時間が与えられましたが、認証機関の容量を申請するための期限と制限が定められていました。
認証機関の立場になってみましょう。認証機関は、クラス D メーカーをサポートし、IVDR に基づいて複雑な適合プロセスを管理するために、適切なタイムラインにわたって予想される作業負荷を考慮する必要があります。したがって、認証機関は、これらの期限を守るメーカーのみが IVDR のより長い移行スケジュールの恩恵を受けることができると伝えています。遅延すると、提出物がレビューキューの最後に追加されることになり、適合性評価が遅れるリスクに直面します。
クラス D IVD の製造業者に対するこれらの期限は 2025 年 5 月に始まり、IVDR 準拠の QMS を整備し、認証機関に IVDR 申請を提出し、(2025 年 9 月に) 認証機関と正式な書面による契約に署名する義務があります。現在利用可能な認証機関の能力を最大限に活用するには、これらのスケジュールを適切に準備して遵守することが賢明です。
IVDR への移行プロジェクトは、特に必要なレベルの技術文書や QMS を初めて使用する場合、どれほど困難で時間がかかるかを承知しています。適切な準備と効率は重要ですが、IVD デバイスを EU 市場に残しておきたい場合は、すべてのメーカーが今すぐ行動を起こす時期であることを認識する必要があります。
Pure Global は、デバイスクラスの確認から、技術文書の提出、QMS の実装、認証機関とのやり取りまで、カスタマイズされたサポートを提供します。当社のIVDR コンサルティング サービスの詳細をご覧ください。



どこにいても、
ご相談ください。
詳しい情報が必要な段階でも、協業を始める段階でも、規制対応の各ステップをサポートします。
お問い合わせ