
Prove cliniche sotto IVDR: qual è un “livello adeguato” per i produttori di IVD?
La conformità sotto la IVDR richiede un approccio rigoroso alla qualità delle prove cliniche. In questo articolo, il Dr. Oliver Eikenberg spiega l'importanza dei processi di valutazione delle prestazioni e della documentazione per garantire adeguate prove cliniche per la conformità dell'UE IVDR.
L'evidenza clinica non è un nuovo requisito nel Regolamento In Vitro Diagnostic Devices (EU IVDR 2017/746); tuttavia, è più chiaramente definito in contrasto con il IVDD. L'articolo 56 e l'allegato XIII della IVDR definiscono le prove cliniche come un processo continuo di valutazione delle prestazioni (PE) per IVD che include validità scientifica, prestazioni analitiche e prestazioni cliniche. In termini più semplici, le prove cliniche comprendono i dati e i risultati della valutazione delle prestazioni per dimostrare che il vostro IVD è sicuro ed esegue secondo il suo scopo previsto.
Considerando i criteri ampliati della IVDR per lo scopo previsto e introducendo un approccio più rigoroso alla sicurezza in generale, molti produttori della IVDR si chiedono fino a che punto devono andare per garantire che essi presentino prove cliniche adeguate, soprattutto quando si avvicinano i termini di transizione IVDR.
Tuttavia, molti produttori di IVD stanno ancora sottovalutando la necessità di un attento processo PE così come i benefici di fornire dati PE chiaramente valutati e analizzati e un adeguato livello di informazioni ai corpi notificati (NB). Poiché le prove cliniche sono i criteri decisionali generali per un dispositivo sicuro ed efficace, la documentazione PE accurata, il processo e l'analisi accurata basata sui rischi sono le chiavi per passare una recensione NB.
Cos'è la valutazione delle prestazioni sotto IVDR?
Valutazione delle prestazioni (PE) è il meccanismo che supporta le prove cliniche. Dovrebbe essere un processo ben strutturato, trasparente, iterativo e continuo attraverso il quale i dati vengono valutati e analizzati per dimostrare la validità scientifica, le prestazioni analitiche e le prestazioni cliniche del dispositivo per il suo scopo previsto. Deve essere parte del QMS del produttore e si verifica durante il ciclo di vita completo compreso il post-mercato.
Come molti altri processi, PE inizia con la pianificazione. Il Piano di valutazione delle prestazioni (PEP) deve definire i metodi di raccolta dei dati, le linee temporali, i criteri di accettazione e le responsabilità del personale. La raccolta di dati PE da test PE viene poi condotta secondo il PEP. I dati scientifici o PE disponibili vengono raccolti e valutati (valutati) in base all'analisi dei dati definita, le cui conclusioni informeranno il Rapporto di valutazione delle prestazioni (PER). Le attività post-market coprono il monitoraggio continuo e la valutazione dello stato dell'arte (ad esempio, standard,Seguito delle prestazioni del mercato post (PMPF), attività documentate nel Rapporto PMPF del produttore e Relazione periodica sull'aggiornamento della sicurezza (PSUR), come applicabile. Risk Management è il nucleo di tutte queste attività e dovrebbe essere incluso in ogni fase.
MDCG 2022-2 illustra il processo PE in un grafico semplificato:
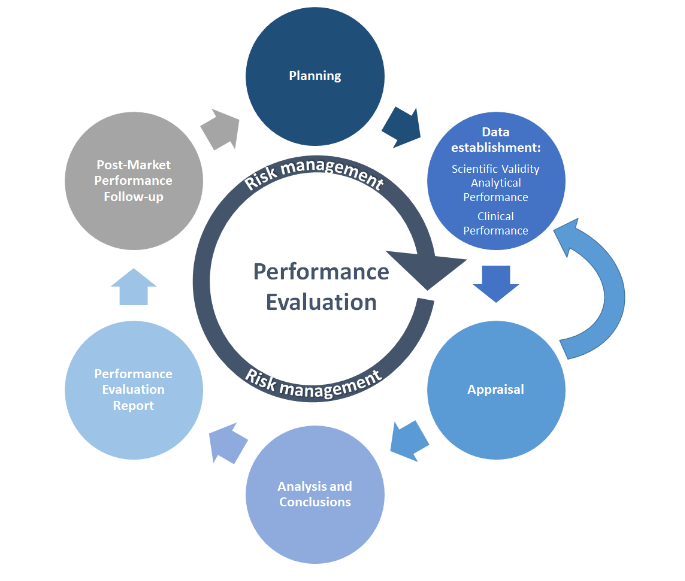
fonte:MDCG 2022-2
Come specificato, le prove cliniche per i IVD possono essere stabilite attraverso la raccolta di dati PE dalla validità scientifica, dalle prestazioni analitiche e dalle prestazioni cliniche. Le prove devono essere raccolte e documentate a un livello che consente una valutazione di documentazione tecnica qualificata (TDA by Notified Body) della sicurezza del dispositivo e se raggiunge i vantaggi clinici previsti quando utilizzati come previsto dal produttore. Questo non significa necessariamente che gli studi clinici sulle prestazioni devono essere condotti. A volte, la valutazione della validità scientifica attraverso valutatori qualificati e test analitici PE con un numero adeguato di campioni clinici sono sufficienti.
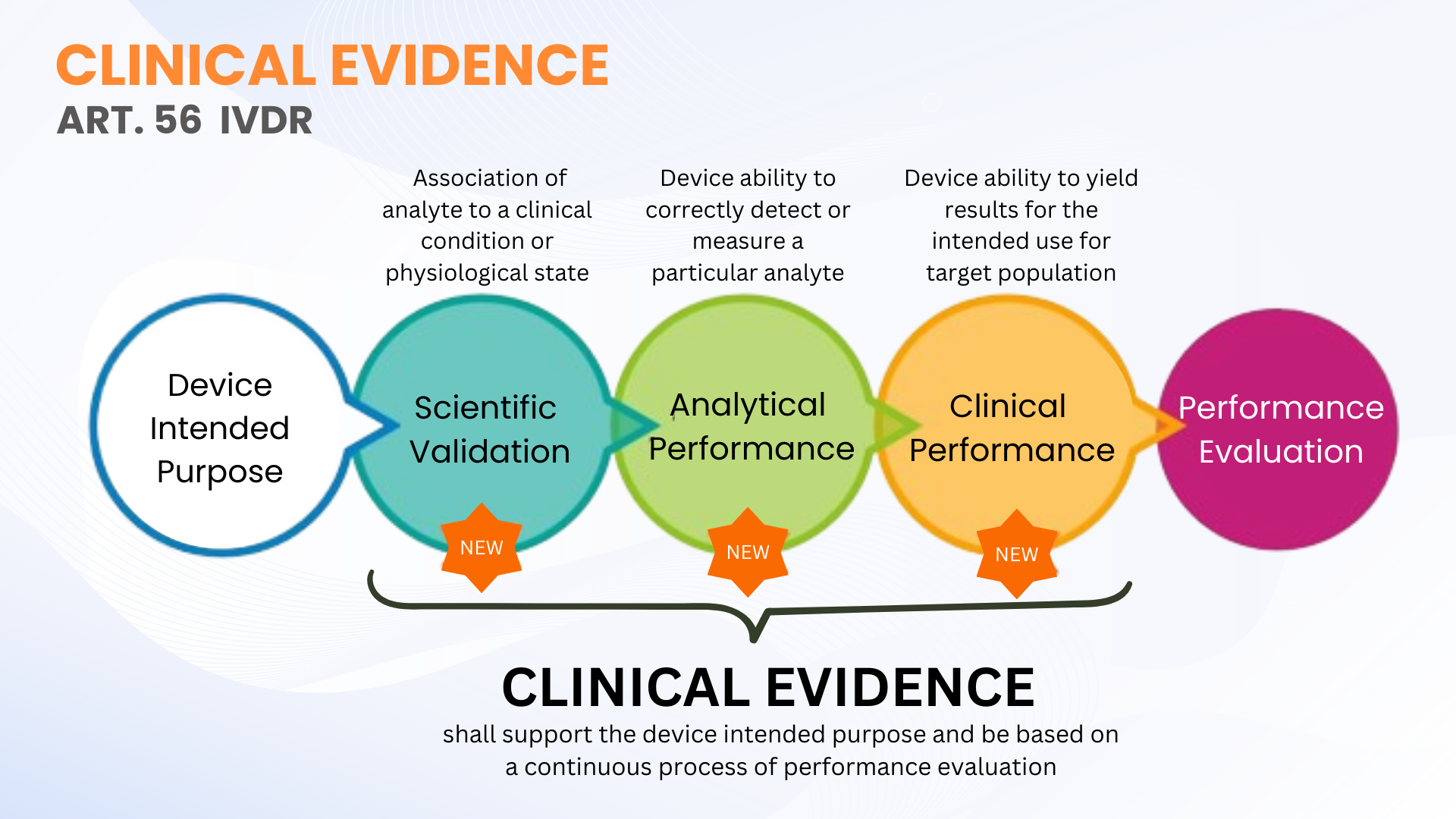
La IVDR non definisce i numeri specifici di campioni clinici da testare per alcuni dispositivi IVD (ad eccezione di alcuni parametri IVD ad alto rischio, dove questi sono definiti nelle specifiche comuni). Questa flessibilità consente ai produttori IVD di pianificare e condurre i test PE appropriati per il loro dispositivo. Tuttavia, i produttori dovrebbero pianificare di scalare il loro approccio di studio delle prestazioni cliniche per sostenere le prove cliniche secondo la classificazione del rischio del dispositivo. Più alta è la classe di rischio, più probabilmente è che gli studi clinici di performance sono necessari per sostenere le prove cliniche.
L'importanza della documentazione PE e della verifica
I produttori IVD devono definire e presentare un PEP in conformità con IVDR, Allegato XIII, parte A, sezione 1.1 prima che effettuano test PE, in quanto il mancato funzionamento potrebbe compromettere l'integrità dei loro dati di prova. Senza prove che i loro test PE hanno seguito il PEP originale (e i criteri di accettazione predefiniti), i recensori NB possono concludere che il PEP è stato rivisto dopo il fatto di giustificare i risultati del test. Gli auditori NB possono facilmente verificare e monitorare quando i PEP sono stati rilasciati, come sono stati seguiti, e quando il test PE è stato fatto. Se questa linea temporale non è chiara, di solito solleva domande, come ad esempio: il processo PE non è chiaramente definito? Perché non è documentato come richiesto dalla IVDR? Perché non sono inclusi criteri di accettazione e metodi per l'analisi dei dati PE? Pertanto, per rendere il vostro design e sviluppo, e il processo di marcatura CE più efficiente, le aziende IVD dovrebbero seguire rigorosamente i processi e i requisiti di documentazione PE ben definiti nella IVDR.
Molti produttori IVD lottano per implementare un processo PE strutturato ma agile ed efficiente che non limita l'innovazione dalle attività di Design & Development. Un semplice requisito di avere una procedura QMS che descrive le loro attività PE o di avere un piano PE chiaramente definito prima di iniziare le attività PE è ancora una sfida importante per molte aziende. Ciò è spesso legato a processi QMS inadeguati, formazione e comprensione dei requisiti PE definiti nella IVDR e linee guida di interpretazione, come MDCG 2022-2.
La relazione PE deve essere approfondita
La documentazione adeguata dei risultati, delle conclusioni e delle razionali del PER è fondamentale per il successo del TDA del produttore in quanto i NB-auditor non sono autorizzati a interpretare alcun dato per conto del produttore. Se la documentazione e i razionali non sono chiaramente espressi, questo potrebbe portare a una stima peggiore da parte dei recensori NB con la necessità di condurre ulteriori test clinici di prestazione o studi PMPF. Un buon metodo per controllare l'adeguatezza è quello di chiedersi se rilasciare il dispositivo IVD in base ai dati PE e alle prove oggettive dimostrate nel vostro PEP e PER. Se non avete le capacità in casa, i valutatori clinici qualificati possono sostenere la scrittura finale del vostro PER per garantire che soddisfi i requisiti dell'auditor NB.
I dispositivi legacy potrebbero avere bisogno di nuove prove cliniche per sostenere uno scopo previsto riveduto
I produttori di IVD legacy che lavorano con uno scopo riveduto e o ampliato previsto nell'ambito della IVDR potrebbero aver bisogno di ridefinire le loro attività PE complessive, la validità scientifica, le prestazioni analitiche e i test clinici delle prestazioni per dimostrare le prove cliniche. In alcuni casi, il PEP potrebbe aver bisogno di essere creato retrospettivamente, se il processo PE non è stato ancora stabilito al momento in cui il dispositivo è stato progettato e sviluppato. Se è così, i produttori dovrebbero descrivere chiaramente questo approccio retrospettivo nel loro processo PE e riscrivere il PEP e PER in conformità con i requisiti IVDR e gli standard all'avanguardia. Tipicamente, gli aggiornamenti sostanziali al PEP e PER richiedono giustificazioni dettagliate e valutazioni dei rischi per gli adattamenti e le modifiche condotte. Se questo non è fatto, i produttori possono affrontare domande significative da recensori NB.
Molti di questi scenari possono essere facilmente evitati se le informazioni PE sono di qualità superiore ed è chiaramente documentato e verificato. L'attuazione dei raccolti Dati di sorveglianza post-market (PMS) può sostenere ulteriormente le prove cliniche come una specifica tecnologia o marcatore. I dati PMS possono anche verificare che la tecnologia o il dispositivo sia ben consolidato e che sia stato ben controllato dal produttore. Questo riduce ulteriormente il rischio complessivo per il dispositivo, che contribuisce al successo della tua recensione NB.
Se avete bisogno di supporto per la creazione di processi PE,aggiornare il QMS a IVDR, o contribuire a scrivere documentazione PE,Pure Global può aiutare. I nostri strumenti software SMART semplificano la raccolta e la valutazione dei dati scientifici e selezionano dispositivi simili o equivalenti sul mercato UE. Scopri di più sul nostro supporto per la valutazione delle prestazioni.



Parliamo,
Ovunque tu sia.
Se siete alla ricerca di maggiori informazioni o pronti a collaborare con noi, siamo qui per guidarvi attraverso ogni fase del processo normativo.
Contattaci