
Preuves cliniques en vertu du RIV: qu'est-ce qu'un niveau adéquat pour les fabricants de DIV?
La conformité en vertu du RIV exige une approche rigoureuse de la qualité des preuves cliniques. Dans cet article, le Dr Oliver Eikenberg explique l'importance des processus d'évaluation du rendement et de la documentation pour assurer une preuve clinique adéquate de la conformité au RIV de l'UE.
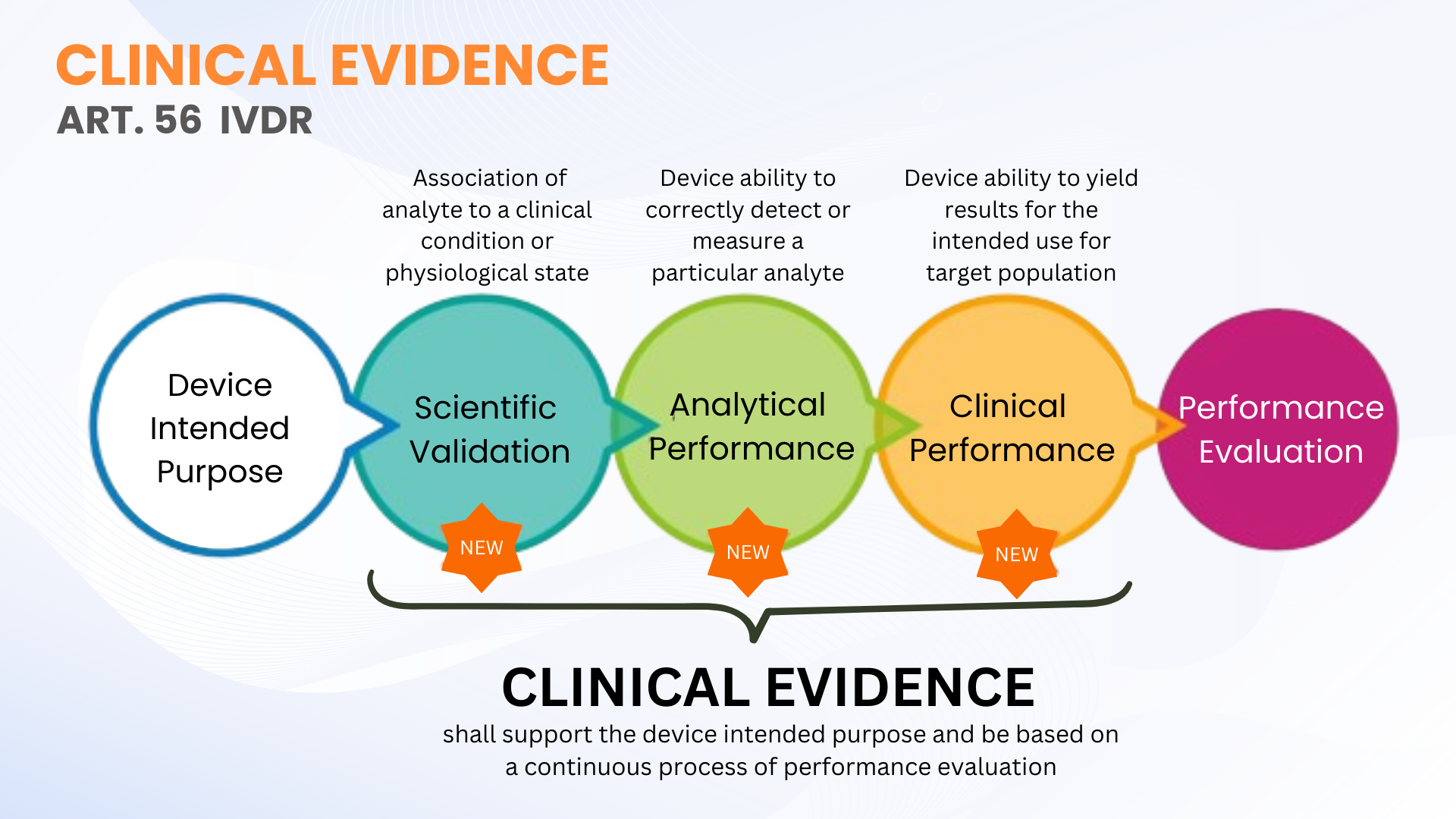
La preuve clinique n'est pas une nouvelle exigence en vertu du règlement sur les dispositifs de diagnostic in vitro (UE IVDR 2017/746); toutefois, elle est plus clairement définie par rapport à la DIV. L'article 56 et l'annexe XIII du RIV définissent les preuves cliniques comme un processus continu de évaluation du rendement (EP) pour les DIV qui comprend la validité scientifique, la performance analytique et la performance clinique. En termes plus simples, les preuves cliniques comprennent les données et les résultats de l'évaluation du rendement afin de démontrer que votre DIV est sécuritaire et qu'elle fonctionne conformément à son objectif.
Compte tenu de l'élargissement des critères de l'IVDR aux fins prévues et de l'adoption d'une approche plus rigoureuse de la sécurité en général, de nombreux fabricants de l'IVDR se demandent jusqu'où ils doivent aller pour s'assurer qu'ils présentent des preuves cliniques adéquates, en particulier à l'approche des délais de transition de l'IVDR.
Cependant, de nombreux fabricants de DIV sous-estiment encore la nécessité d'un processus de PE prudent ainsi que les avantages de fournir des données de PE clairement évaluées et analysées et un niveau d'information adéquat aux organismes notifiés (NB). Étant donné que les preuves cliniques constituent les critères de décision généraux pour un dispositif sûr et efficace, une documentation complète de PE, un processus et une analyse précise des données fondées sur le risque sont les clés pour réussir un examen du NB.
Qu'est-ce que l'évaluation du rendement dans le cadre de l'IVDR?
L'évaluation du rendement (EP) est le mécanisme qui appuie vos données cliniques. Il devrait s'agir d'un processus bien structuré, transparent, itératif et continu par lequel les données sont évaluées et analysées afin de démontrer la validité scientifique, la performance analytique et la performance clinique de l'appareil aux fins prévues. Il doit faire partie du SGQ du fabricant et se produit tout au long du cycle de vie, y compris après la commercialisation.
Comme beaucoup d'autres processus, PE commence par la planification. Le plan d'évaluation du rendement (PEP) doit définir les méthodes de collecte des données, les échéanciers, les critères d'acceptation et les responsabilités du personnel. La collecte de données PE à partir des essais PE est ensuite effectuée selon la PEP. Les données scientifiques ou PE disponibles sont recueillies et évaluées (évaluations) sur la base de l'analyse des données définies, dont les conclusions éclaireront le rapport d'évaluation de la performance (PER). Les activités post-commercialisation couvrent le suivi et l'évaluation continus de l'état de la technique (par exemple, les normes,Suivi de la performance après le marché, les activités documentées dans le rapport PMPF du fabricant et le rapport périodique de mise à jour de sécurité (PSUR), selon le cas). La gestion du risque est au cœur de toutes ces activités et devrait être incluse à chaque étape.
MDCG 2022-2 illustre le processus de PE dans un graphique simplifié:
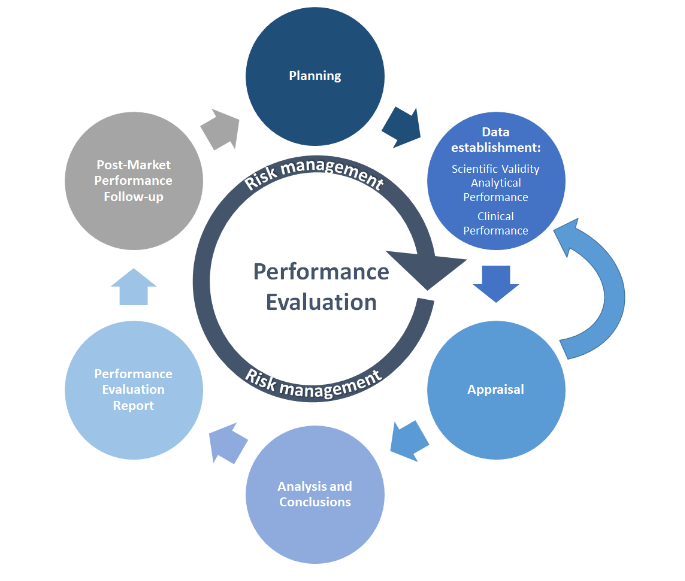
Source:MDCG 2022-2
Comme nous l'avons indiqué, des données cliniques sur les MIV peuvent être établies grâce à la collecte de données de PE à partir de la validité scientifique, du rendement analytique et du rendement clinique. Les preuves doivent être recueillies et documentées à un niveau qui permet une évaluation de la documentation technique qualifiée (AMT par Notified Body) de la sécurité du dispositif et si celui-ci permet d'obtenir les avantages cliniques escomptés lorsqu'il est utilisé comme prévu par le fabricant. Cela ne signifie pas nécessairement que des études de performance clinique doivent être menées. Parfois, l'évaluation de la validité scientifique par l'entremise d'évaluateurs qualifiés et de tests analytiques de PE avec un nombre suffisant d'échantillons cliniques est suffisante.
Le RIV ne définit pas le nombre précis d'échantillons cliniques à tester pour certains dispositifs de DIV (sauf pour certains paramètres de DIV à risque élevé, lorsque ceux-ci sont définis dans les spécifications communes). Cette flexibilité permet aux fabricants de DIV de planifier et de réaliser les essais de PE appropriés pour leur appareil. Cependant, les fabricants devraient planifier d'évaluer à l'échelle de leur approche d'étude de la performance clinique afin d'étayer les preuves cliniques selon la classification des risques de l'appareil. Plus la classe de risque est élevée, plus il est probable que des études de performance clinique soient nécessaires pour étayer les données cliniques.
L'importance de la documentation et de la vérification PE
Les fabricants de DIV doivent définir et soumettre une PEP conformément à l'annexe XIII de l'IVDR, partie A, section 1.1 avant ils effectuent des essais d'EP, car le fait de ne pas le faire pourrait compromettre l'intégrité de leurs données d'essai. En l'absence de preuves que leurs essais de PE ont suivi le PEP original (et des critères d'acceptation prédéfinis), les évaluateurs du NB peuvent conclure que le PEP a été révisé après le fait pour étayer les résultats du test. Les auditeurs du NB peuvent facilement vérifier et suivre le moment où les PEP ont été libérées, la façon dont elles ont été suivies et le moment où les tests de PE ont été effectués. Si ce calendrier n'est pas clair, il soulève habituellement des questions, comme: Le processus de PE n'est-il pas clairement défini? Pourquoi n'est-elle pas documentée comme l'exige l'IVDR? Pourquoi les critères et les méthodes d'acceptation pour l'analyse des données PE ne sont-ils pas inclus? Par conséquent, pour rendre votre conception et votre développement plus efficaces et le processus de marquage CE, les entreprises de DIV devraient suivre strictement les processus et les exigences bien définis de documentation PE dans le DIV.
De nombreux fabricants d'IVD peinent à mettre en œuvre un processus PE structuré mais agile et efficace qui ne limite pas l'innovation des activités de conception et de développement. Une simple exigence d'avoir une procédure de SGQ décrivant leurs activités de PE ou d'avoir un plan de PE clairement défini avant de commencer les activités de PE reste un défi majeur pour de nombreuses entreprises. Cela est souvent lié à des processus de SGQ inadéquats, à la formation et à la compréhension des exigences de PE définies dans les lignes directrices de l'IVDR et d'interprétation, comme la MDCG 2022-2.
Les rapports PE doivent être complets.
Une documentation adéquate des résultats, des conclusions et des justifications du PE dans le RAP est essentielle au succès des fabricants, puisque les auditeurs du NB ne sont pas autorisés à interpréter des données au nom du fabricant. Si la documentation et les justifications ne sont pas clairement exprimées, cela pourrait entraîner une estimation du pire cas par les évaluateurs du NB, avec la nécessité d'effectuer des essais cliniques supplémentaires ou des études sur le PMPF. Une bonne méthode pour vérifier l'adéquation est de vous demander si vous relâchez votre appareil d'IVD en fonction des données PE et des preuves objectives démontrées dans votre PPE et PER. Si vous n'avez pas les capacités internes, des évaluateurs cliniques qualifiés peuvent appuyer la rédaction finale de votre RAP pour s'assurer qu'il répond aux exigences du vérificateur du Nouveau-Brunswick.
Les dispositifs hérités pourraient avoir besoin de nouvelles preuves cliniques pour appuyer un objectif révisé
Les fabricants de DIV hérités travaillant avec un objectif révisé ou élargi dans le cadre du DIV pourraient avoir besoin de redéfinir leurs activités de PE globale, leur validité scientifique, leur rendement analytique et les tests de performance clinique pour démontrer les preuves cliniques. Dans certains cas, il faudra peut-être créer le PEP rétrospectivement si le processus de PE n'a pas encore été établi au moment de la conception et de la mise au point du dispositif. Dans l'affirmative, les fabricants devraient décrire clairement cette approche rétrospective dans leur processus de PE et réécrire le PEP et le PER conformément aux exigences de l'IVDR et aux normes de pointe. En règle générale, les mises à jour substantielles du PPE et du PER exigent des justifications détaillées et des évaluations des risques pour les adaptations et les changements effectués. Si cela n'est pas fait, les fabricants peuvent faire face à des questions importantes de la part des évaluateurs du NB.
Beaucoup de ces scénarios peuvent être facilement évités si l'information sur les PE est de qualité supérieure et est clairement documentée et vérifiée. Mise en œuvre des mesures Données relatives à la surveillance après la mise en marché peut appuyer davantage les preuves cliniques comme une technologie ou un marqueur spécifique. Les données du SPM peuvent également vérifier que la technologie ou l'appareil est bien établi et qu'il a été bien contrôlé par le fabricant. Cela réduit encore le risque global pour l'appareil, ce qui contribue au succès de votre examen du NB.
Si vous avez besoin d'aide pour établir des processus de PE,la mise à jour de votre QMS à IVDR, ou aider à rédiger la documentation PE,Pure Global peut aider. Nos outils logiciels SMART simplifient la collecte et l'évaluation des données scientifiques ainsi que la sélection d'appareils similaires ou équivalents sur le marché de l'UE. En savoir plus sur notre appui à l ' évaluation des résultats.



Parlons,
N'importe où vous êtes.
Que vous cherchiez plus d'information ou que vous soyez prêt à travailler en partenariat avec nous, nous sommes là pour vous guider à chaque étape du processus réglementaire.
Contactez-nous