
IVDR に基づく認証機関との連携: 申請前プロセスについてレガシー メーカーが知っておくべきこと
IVDR 移行には、IVD メーカーが認証機関と連携するために従う必要がある構造化プロセスが含まれています。多くの IVD メーカーは初めて認証機関とやり取りするため、パートナーシップを確実に成功させるには何が必要かを理解する必要があります。この記事では、オリバー アイケンバーグ博士が、製造業者が何を準備しなければならないか、また IVDR CE マーキングのためにいつ認証機関と連携する必要があるかを明確にしています。
(EU) 2017/746 (IVDR) に基づく CE マーキングを取得するには、クラス A 無菌、クラス B、C、または D に分類される IVDs の合法的な製造業者は、認定認証機関 (NB) との正式な申請書に署名する必要があります。このプロセスに精通している人であれば、NB を選択し、NB ごとに異なる可能性がある NB アプリケーションの段階に従うことの複雑さを理解していると思います。 IVDR 移行要件に沿った NB の申請期限が迫っているため、IVDR 企業は最終的な IVDR 期限までに NB の事前申請プロセスを理解する必要があります。これは、IVDs をレガシー デバイスとして販売し、独立した NB レビュー担当者と初めて関わる IVD メーカーにとって特に重要です。
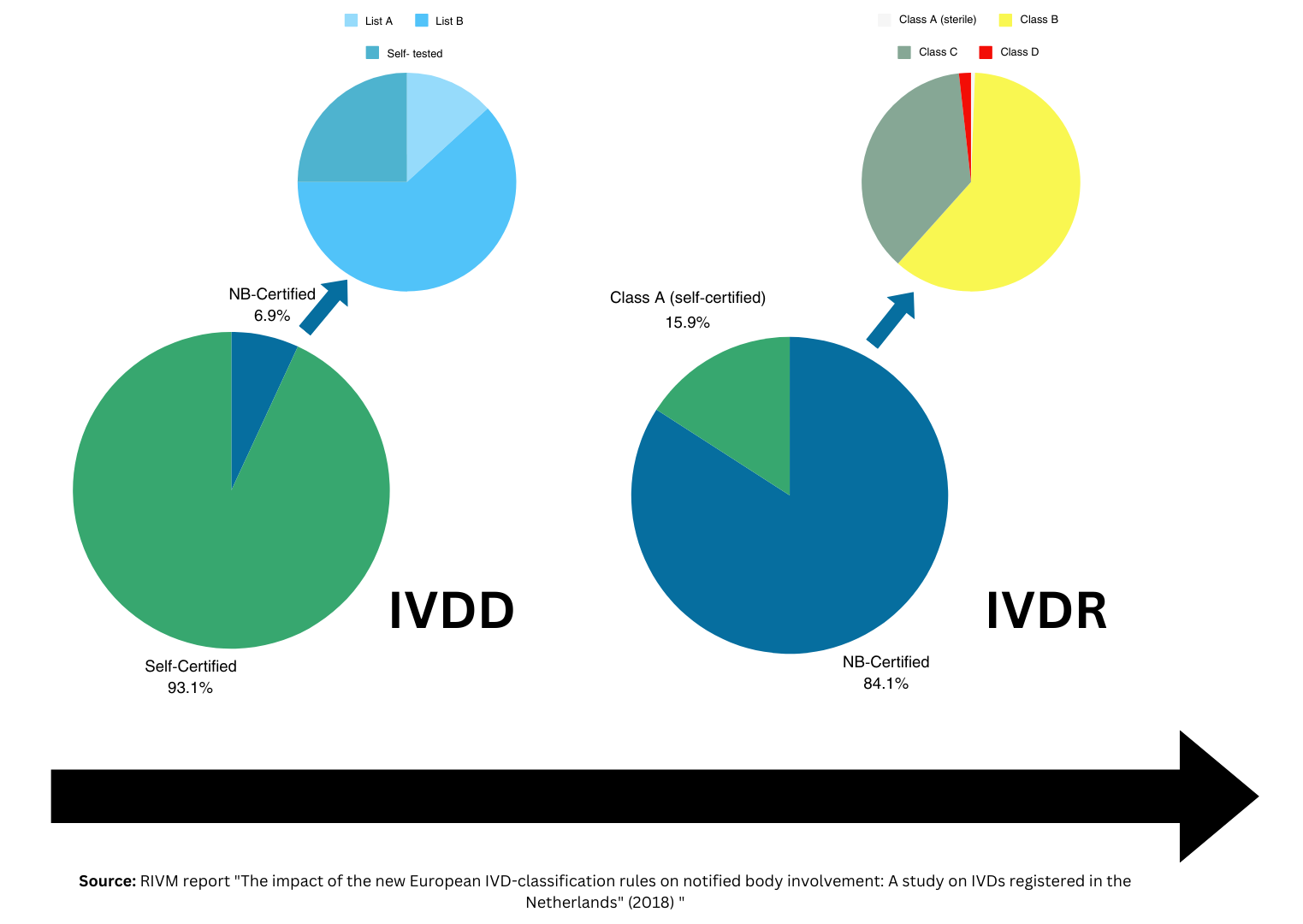
レガシー デバイス メーカーは、IVDR 規制の最新の修正を認識する必要があります。 EU 2024/1860では、一連の重要なアップデートが紹介されています。これには、EUDAMED の段階的導入、新しいサプライ チェーンの義務、従来の IVD デバイスに対する次のような改訂された移行規定が含まれます。
EU 市場内で事業を継続したいすべての IVD メーカーにとって、この修正の制限、期限、および特定の要件を理解することが不可欠です。従来の IVD デバイスの場合、新しい期限と申請前のさまざまな手順が含まれるため、慎重に検討する必要があります。
IVD メーカー向けの公認機関の事前申請プロセス
Team-NB のメンバーは、医療機器メーカーの申請前、申請、申請後のプロセスを詳しく説明したMDR 認定に関する合意文書を発行しました。この合意文書は、MDR に移行するレガシー医療機器 (第 120 条に基づく) と、新しく市場に投入され、これまで指令に基づいて認定されていない医療機器に適用されます。これは拘束力のある文書ではなく、IVDs 向けにまだ公開されていませんが、「付録 A: プロセスのさまざまな段階でメーカーが提出するデータ/文書のリスト」に示されている NB の一般的な管理と義務の詳細を示しています。アプリケーション段階の全体的なアプローチは、おそらく IVDs と同じです。
NB との最初のやり取りには、申請書の提出と NB との契約への署名が含まれます (通常は 4 か月後)。これらの手順は、NB が技術文書評価 (TDA) レビューを実行するために完全なデータを要求する数か月前に完了する必要があります。準備段階により、NB は内部リソースのバランスをとり、最終移行期限が終了する前にタイムリーなサービス (レビュー) を提供できるようになります。
IVD メーカーが NB に申請を提出するには何が必要ですか?
MDR または IVDR が草案されたときにこれらの段階が考慮されていなかったため、前段階の TDA レビュー プロセスに関する詳細な義務や詳細は存在しません。ただし、EU 委員会は、解釈の目的で規則 (EU) 2024/1860 によって修正された、IVDR に規定されている延長移行期間の実施に関連する実際的な側面に関する Q&Aを公開しています。
この Q&A は、申請書を提出する前、または書面による契約書に署名する前に、NB が申請書の完全なレビュー (完全な TDA) を行う必要がないことを明確にしています。 IVD メーカーが技術文書を改良するにはさらに時間がかかる可能性があるため、これは合理的です。また、申請書は、NB が製品を IVDs として認定し、そのIVDR による分類を識別し、選択した適合性評価手順を特定できるようにする必要があることも明確にしています。申請書を提出する際、製造業者は予定される提出のスケジュールを提供する必要があります。これらすべての情報は、IVD メーカーがアプリケーションを NB に提出する前に IVDR の主要な要素を理解し、実装していることを示しています。
分類と使用目的
分類は、IVDR 付録 20.4.1 (c) の基準に従ってラベルに意図された目的が明確に表現されている場合にのみ確認できるため、出願前のリクエストで頻繁に問題になります。客観的な証拠を得るには、使用説明書 (IFU) とラベルに意図された目的を反映する必要があります。メーカーが IVDs を IVDD で販売し続けることができる場合でも、新しい使用目的を準備し、管理された文書で提案された IFU の草案を作成することを強くお勧めします。そうしないと、分類を確認できず、申請書は NB によって署名されません。このトピックについてさらに詳しく知りたい場合は、IVDR に基づく本来の目的の役割に関するブログをご覧ください。
QMS 出力
チーム NB コンセンサス文書の付録 A によると、SRN 番号、UDI-DI、EMDN コード、選択したEU 正式な代表者 (EU REP)、および関係する「重要なサプライヤーおよび/または重要な下請け業者」に関する情報も事前申請段階で必要となります。これらは、すべての IVD 製造業者が 2025 年 5 月 26 日までに実装する必要がある基本的な QMS 要件からの出力です。ただし、IVD 製造業者が QMS 手順を積極的に理解し、時間内に完了しない場合、NB に必要な情報の出力は適切ではなくなります。たとえ IVD メーカーが数か月後に NB による監査を受けたとしても、2025 年 5 月 26 日までに IVDR 対応の QMS の証拠が欠けている場合は、依然として重大な不適合となる可能性があります。 QMS の内部では、NB がいつ、何が実装され、有効になったかを簡単に特定できることに注意してください。
技術文書
特に、チーム NB のコンセンサス文書には、NB が申請の一部として、概要/セクションまたは完全な技術文書を要求する可能性があると記載されています。これは、デバイスとしての資格、それぞれの分類、および選択された適合性評価手順を検証するために、デバイスに関する十分な情報を収集するためです。この文書にはさらに、「申請の審査に基づいて、NBは申請を受理するか拒否するかを決定する(契約締結後にのみ)」と記載されている。これは、NB があなたの申請を受け付けない可能性があり、その場合は別の NB に申請するかどうかはあなた次第であることを意味します。これにより、特に NB の申請期限がすでに過ぎている場合、市場投入までの時間が大幅に変わる可能性があります。
IVDR 申請前の段階では遵守が不可欠です
結論として、IVDR の多くの要素は、NB への申請が提出され、NB との契約が署名される少し前に理解され、適切な方法で実装される必要があります。これは、IVDR 第 10 条で取り上げられている意図された目的、分類、および基本的な QMS 手順(EUDAMED 登録、ラベル付け、UDI、PMS、PE プロセス、およびビジランスを含む)に特に当てはまります。 NB は、IVDR の要件に従って QMS の手順にどのように従ったかを証明する文書を要求する場合があることに注意してください。製造業者は、安全性、ラベル表示、リスク管理など、最近発行された最先端の基準と解釈に従ってプロセスと基本要件に従う必要があります。たとえば、IVD の新しいシンボルやラベル情報は、適用される安全規制や最新の EMDN コードに沿った分類への適切な準拠を示します。
申請書を提出したり契約書に署名したりするための NB プロセス中にどのような詳細な質問が生じる可能性があるかを予測するには、移行スケジュールが早すぎます。ただし、IVD メーカーは、潜在的な不適合や申請の拒否を避けるために、できるだけ早く IVDR の要件に照らして自社の文書を積極的にチェックする必要があります。
Pure Global の経験豊富な IVDR 専門家が、認証機関の検索と選択および申請前の段階をお手伝いします。 IVDR 分類の確認、QMS プロセスの実装、文書化とラベル付けのレビューなどを支援します。 私たちがどのようにお手伝いできるかについては、お問い合わせください。



どこにいても、
ご相談ください。
詳しい情報が必要な段階でも、協業を始める段階でも、規制対応の各ステップをサポートします。
お問い合わせ