
Notícias Regulatórias Semanais: 23 de abril a 1 de maio de 2025
Nesta semana nas notícias regulatórias de dispositivos médicos, o FDA lançou o ESG NextGen, uma plataforma modernizada para peticionamento eletrônico seguro que substitui o sistema legado WebTrader; e a Anvisa do Brasil reclassificou os dispositivos de PCR ultrassensível da Classe de Risco III para a Classe II, simplificando o caminho regulatório de registro para notificação.
ESTADOS UNIDOS
FDA lança ESG NextGen: uma plataforma modernizada para submissões regulatórias eletrônicas seguras
Em 23 de abril de 2025, o FDA apresentou o novo Electronic Submission Gateway Next Generation (ESG NextGen), uma plataforma modernizada e baseada na web projetada para simplificar e tornar mais seguras as submissões regulatórias eletrônicas. Substituindo a interface legado WebTrader, o Unified Submission Portal (USP) permite que os usuários enviem e acompanhem informações regulatórias com maior eficiência e melhor experiência de usuário. O sistema ESG original foi totalmente descontinuado. Guias de usuário e recursos de treinamento estão disponíveis para auxiliar na transição.
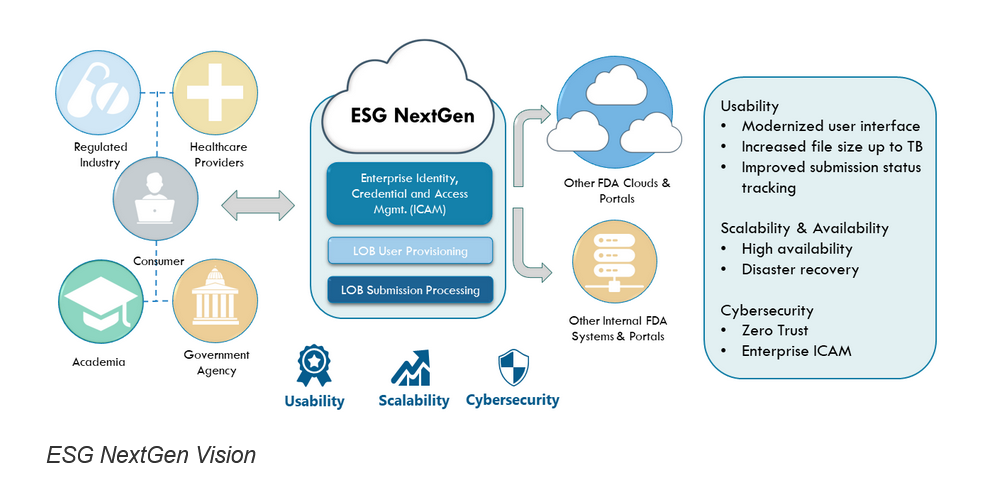
🔗 Gateway de Submissões Eletrônicas | FDA
Saiba mais sobre o Registro de Dispositivos Médicos no FDA dos EUA.
BRASIL
Diretrizes revisadas sobre enquadramento sanitário para dispositivos de PCR ultrassensível
A Anvisa revisou as diretrizes de enquadramento sanitário para dispositivos médicos que medem o parâmetro de PCR (proteína C-reativa) ultrassensível. Como resultado dessa mudança, esses dispositivos foram reclassificados da Classe de Risco III para a Classe de Risco II, o que significa que agora estão sujeitos a notificação em vez de registro.
Esta atualização baseia-se na Nota Técnica 17/2025/SEI/GEVIT/GGTPS/DIRE3/ANVISA e alinha-se com as disposições da Resolução da Diretoria Colegiada (RDC) 830/2023, que rege a notificação e o registro de dispositivos médicos para diagnóstico in vitro (DIV).
As empresas que já protocolaram solicitações de reclassificação serão contatadas pela Gerência de Produtos para Diagnóstico In Vitro com orientações sobre os próximos passos. Isso também se aplica a quaisquer registros de Classe III protocolados ou concedidos durante a vigência atual da RDC 830/2023.
Para todas as novas submissões relacionadas a testes de PCR ultrassensível, a Classe de Risco II revisada deve ser aplicada, refletindo seu status atualizado como produtos sujeitos a notificação.
O teste de PCR ultrassensível desempenha um papel vital na saúde ao monitorar e avaliar o risco de eventos cardiovasculares. Ele permite a detecção precoce de inflamação e de infecções vasculares em indivíduos que podem ainda não apresentar sintomas clínicos.
🔗 Para mais informações, visite: Diretrizes sobre enquadramento sanitário para o parâmetro de PCR ultrassensível
Saiba mais sobre as regulamentações de dispositivos médicos no Brasil.
 MercadoRegistro de Dispositivos Médicos na FDA dos EUAMercadoRegistro de Dispositivos Médicos na ANVISA BrasilArtigo do BlogComo registrar dispositivos médicos e DIVs no BrasilRelatório de PesquisaMonitores Contínuos de Glicose: O Mapa Regulatório GlobalRelatório de PesquisaIA como Dispositivo Médico: O Mapa Regulatório GlobalArtigo do BlogTransforme Aprovações da FDA & EU em Acesso ao Mercado GlobalVídeoComo registrar um dispositivo médico na FDA dos EUAVídeoUDI no Brasil: Insights para Fabricantes de Dispositivos MédicosVídeoComo Registrar um Dispositivo Médico no BrasilServiço de MercadoAgente da US FDA para Empresas de Dispositivos Médicos
MercadoRegistro de Dispositivos Médicos na FDA dos EUAMercadoRegistro de Dispositivos Médicos na ANVISA BrasilArtigo do BlogComo registrar dispositivos médicos e DIVs no BrasilRelatório de PesquisaMonitores Contínuos de Glicose: O Mapa Regulatório GlobalRelatório de PesquisaIA como Dispositivo Médico: O Mapa Regulatório GlobalArtigo do BlogTransforme Aprovações da FDA & EU em Acesso ao Mercado GlobalVídeoComo registrar um dispositivo médico na FDA dos EUAVídeoUDI no Brasil: Insights para Fabricantes de Dispositivos MédicosVídeoComo Registrar um Dispositivo Médico no BrasilServiço de MercadoAgente da US FDA para Empresas de Dispositivos MédicosVamos conversar,
Onde quer que você esteja.
Quer esteja procurando mais informações ou pronto para ser nosso parceiro, estamos aqui para orientá-lo em todas as etapas do processo regulatório.
Fale conosco