
Evidência clínica no IVDR: “nível adequado” a fabricantes de DIV
A conformidade sob o IVDR exige uma abordagem rigorosa em relação à qualidade da evidência clínica. Neste artigo, o Dr. Oliver Eikenberg explica a importância dos processos de avaliação de desempenho e da documentação para garantir evidência clínica adequada para a conformidade com o IVDR da UE.
A evidência clínica não é um requisito novo sob o Regulamento de Dispositivos Médicos para Diagnóstico In Vitro (EU IVDR 2017/746); no entanto, ela está definida de forma mais clara em contraste com a IVDD. O Artigo 56 e o Anexo XIII do IVDR definem a evidência clínica como um processo contínuo de avaliação de desempenho (PE) para IVDs que inclui validade científica, desempenho analítico e desempenho clínico. Em termos mais simples, a evidência clínica compreende os dados e as conclusões da avaliação de desempenho para demonstrar que seu IVD é seguro e funciona de acordo com a finalidade prevista.
Considerando que o IVDR expandiu os critérios para a finalidade prevista e introduziu uma abordagem mais rigorosa para a segurança em geral, muitos fabricantes sob o IVDR estão se perguntando até onde precisam ir para garantir a apresentação de evidências clínicas adequadas, especialmente com a aproximação dos prazos de transição do IVDR.
No entanto, muitos fabricantes de IVDs ainda estão subestimando a necessidade de um processo cuidadoso de PE, bem como os benefícios de fornecer dados de PE claramente avaliados e analisados e um nível adequado de informações aos Organismos Notificados (NBs). Como a evidência clínica é o critério de decisão geral para um dispositivo seguro e eficaz, uma documentação de PE minuciosa, o processo e a análise precisa de dados baseada em risco são as chaves para passar por uma revisão de NB.
O que é a avaliação de desempenho sob o IVDR?
A avaliação de desempenho (PE) é o mecanismo que dá suporte à sua evidência clínica. Ela deve ser um processo bem estruturado, transparente, iterativo e contínuo pelo qual os dados são avaliados e analisados para demonstrar a validade científica, o desempenho analítico e o desempenho clínico do dispositivo para a sua finalidade prevista. Ela deve fazer parte do QMS do fabricante e ocorrer ao longo de todo o ciclo de vida, incluindo o pós-mercado.
Como muitos outros processos, a PE começa com o planejamento. O Plano de Avaliação de Desempenho (PEP) deve definir os métodos de coleta de dados, cronogramas, critérios de aceitação e as responsabilidades do pessoal. A coleta de dados de PE a partir dos testes de PE é então realizada de acordo com o PEP. Os dados científicos ou de PE disponíveis são coletados e avaliados (apreciados) com base na análise de dados definida, cujas conclusões subsidiarão o Relatório de Avaliação de Desempenho (PER). As atividades pós-mercado cobrem o monitoramento contínuo e a avaliação do estado da arte (por exemplo, normas, atividades de Acompanhamento de Desempenho Pós-Mercado (PMPF) documentadas no Relatório de PMPF do fabricante e no Relatório Periódico de Atualização de Segurança (PSUR), conforme aplicável). O gerenciamento de riscos é o cerne de todas essas atividades e deve ser incluído em cada etapa.
O MDCG 2022-2 ilustra o processo de PE em um gráfico simplificado:
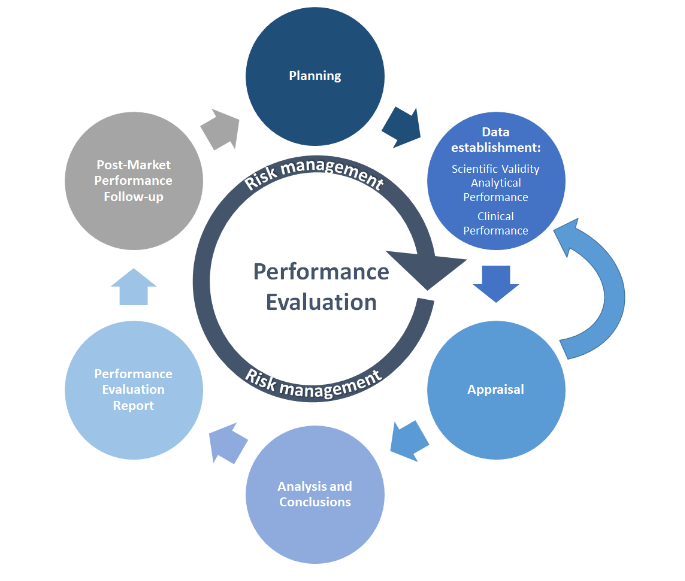
fonte: MDCG 2022-2
Como mencionado, a evidência clínica para IVDs pode ser estabelecida por meio da coleta de dados de PE a partir da validade científica, do desempenho analítico e do desempenho clínico. A evidência deve ser coletada e documentada em um nível que permita uma avaliação qualificada da documentação técnica (TDA, realizada pelo Organismo Notificado) sobre a segurança do dispositivo e se ele alcança os benefícios clínicos pretendidos quando utilizado conforme previsto pelo fabricante. Isso não significa necessariamente que estudos de desempenho clínico devam ser realizados. Às vezes, a avaliação da validade científica por meio de avaliadores qualificados e testes analíticos de PE com um número adequado de amostras clínicas são suficientes.
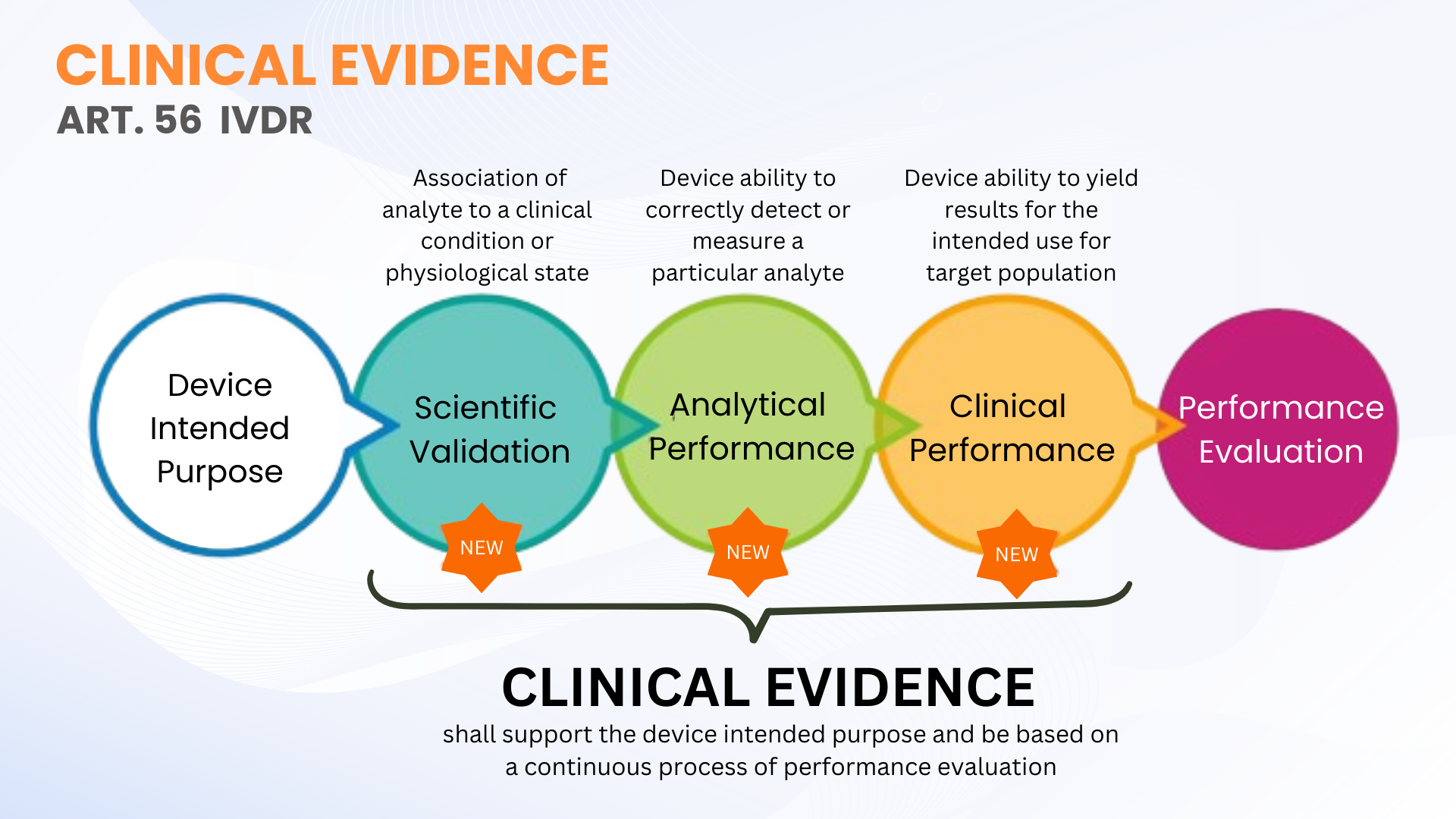
O IVDR não define números específicos de amostras clínicas a serem testadas para determinados dispositivos IVD (exceto para alguns parâmetros de IVD de alto risco, onde estes são definidos nas Especificações Comuns). Essa flexibilidade permite que os fabricantes de IVDs planejem e conduzam os testes de PE adequados para seus dispositivos. No entanto, os fabricantes devem planejar o dimensionamento de sua abordagem de estudo de desempenho clínico para dar suporte à evidência clínica de acordo com a classificação de risco do dispositivo. Quanto maior a classe de risco, maior a probabilidade de que estudos de desempenho clínico sejam necessários para fundamentar a evidência clínica.
A importância da documentação e verificação da PE
Os fabricantes de IVDs devem definir e enviar um PEP de acordo com o IVDR, Anexo XIII, Parte A, Seção 1.1 antes de realizar os testes de PE, pois a falha em fazer isso pode comprometer a integridade de seus dados de teste. Sem evidências de que os testes de PE seguiram o PEP original (e os critérios de aceitação pré-definidos), os revisores dos NBs podem concluir que o PEP foi revisado a posteriori para substanciar os resultados dos testes. Os auditores dos NBs podem facilmente verificar e rastrear quando os PEPs foram emitidos, como foram seguidos e quando os testes de PE foram realizados. Se esse cronograma não for claro, isso geralmente levanta questões, tais como: O processo de PE não está claramente definido? Por que não está documentado conforme exigido pelo IVDR? Por que os critérios de aceitação e os métodos para a análise de dados de PE não foram incluídos? Portanto, para tornar o seu processo de projeto e desenvolvimento, bem como de marcação CE, mais eficiente, as empresas de IVD devem seguir estritamente os processos e requisitos de documentação de PE bem definidos no IVDR.
Muitos fabricantes de IVDs lutam para implementar um processo de PE estruturado, mas ágil e eficiente, que não limite a inovação decorrente das atividades de Projeto e Desenvolvimento. O simples requisito de ter um procedimento de QMS descrevendo suas atividades de PE ou de ter um Plano de PE claramente definido antes de iniciar as atividades de PE ainda é um grande desafio para muitas empresas. Isso geralmente está relacionado a processos de QMS inadequados, falta de treinamento e falta de compreensão dos requisitos de PE definidos no IVDR e nas diretrizes de interpretação, como o MDCG 2022-2.
O relatório de PE deve ser detalhado
A documentação adequada dos resultados, conclusões e justificativas de PE no PER é fundamental para o sucesso da TDA do fabricante, uma vez que os auditores dos NBs não têm permissão para interpretar quaisquer dados em nome do fabricante. Se a documentação e as justificativas não forem expressas de forma clara, isso poderá resultar em uma avaliação do pior cenário por parte dos revisores dos NBs, gerando a necessidade de realizar testes adicionais de desempenho clínico ou estudos de PMPF. Um bom método para verificar a adequação é perguntar a si mesmo se você liberaria seu dispositivo IVD com base nos dados de PE e nas evidências objetivas demonstradas em seu PEP e PER. Se você não tiver as competências internas, avaliadores clínicos qualificados podem dar suporte na redação final do seu PER para garantir que ele atenda aos requisitos dos auditores dos NBs.
Dispositivos legados podem precisar de novas evidências clínicas para dar suporte a uma finalidade prevista revisada
Os fabricantes de IVDs legados que trabalham com uma finalidade prevista revisada e/ou expandida sob o IVDR podem precisar redefinir suas atividades gerais de PE, validade científica, desempenho analítico e testes de desempenho clínico para demonstrar a evidência clínica. Em alguns casos, o PEP pode precisar ser criado retrospectivamente, se o processo de PE ainda não estivesse estabelecido no momento em que o dispositivo foi projetado e desenvolvido. Se for o caso, os fabricantes devem descrever claramente essa abordagem retrospectiva em seu processo de PE e reescrever o PEP e o PER em conformidade com os requisitos do IVDR e as normas do estado da arte. Normalmente, atualizações substanciais no PEP e no PER exigem justificativas detalhadas e avaliações de risco para as adaptações e alterações realizadas. Se isso não for feito, os fabricantes poderão enfrentar questionamentos significativos por parte dos revisores dos NBs.
Muitos desses cenários podem ser facilmente evitados se as informações de PE forem de qualidade superior e estiverem claramente documentadas e verificadas. A implementação de dados de Vigilância Pós-Mercado (PMS) coletados pode dar mais suporte à evidência clínica, como no caso de uma tecnologia ou marcador específico. Os dados de PMS também podem verificar se a tecnologia ou o dispositivo está bem estabelecido e se tem sido bem controlado pelo fabricante. Isso reduz ainda mais o risco geral do dispositivo, o que contribui para o sucesso da sua revisão por parte do NB.
Se precisar de suporte para estabelecer processos de PE, atualizar seu QMS para o IVDR ou de ajuda para redigir a documentação de PE, a Pure Global pode ajudar. Nossas ferramentas de software SMART simplificam a coleta e a avaliação de dados científicos, bem como a seleção de dispositivos semelhantes ou equivalentes no mercado da UE. Saiba mais sobre o nosso suporte para avaliação de desempenho.
 MercadoClassificação e Regulamentação de Dispositivos Médicos na UE (MDR/IVDR)Atualização RegulatóriaAtualização de Normas Harmonizadas da UE 2026Atualização RegulatóriaMDCG 2021-5 Rev.1 Transição para o Símbolo EU REPAtualização RegulatóriaRegulamento (UE) 2026/977: regras de prazos e custos para ONsAtualização RegulatóriaTerceira Atualização do Formulário MIR v7.3.1 (SB-11154)VídeoPasso a Passo: Marcação CE para Dispositivos Médicos sob a EU MDRVídeoMDCG 2025-5: Regras do IVDR para Estudos de Desempenho de IVDVídeoMudanças no IVDR e impactos para fabricantes de IVDServiço de MercadoAcompanhamento Clínico Pós-Mercado (PMCF) da UE para Dispositivos MédicosServiço de MercadoAcompanhamento do Desempenho Pós-Mercado (PMPF) da UE para IVDs
MercadoClassificação e Regulamentação de Dispositivos Médicos na UE (MDR/IVDR)Atualização RegulatóriaAtualização de Normas Harmonizadas da UE 2026Atualização RegulatóriaMDCG 2021-5 Rev.1 Transição para o Símbolo EU REPAtualização RegulatóriaRegulamento (UE) 2026/977: regras de prazos e custos para ONsAtualização RegulatóriaTerceira Atualização do Formulário MIR v7.3.1 (SB-11154)VídeoPasso a Passo: Marcação CE para Dispositivos Médicos sob a EU MDRVídeoMDCG 2025-5: Regras do IVDR para Estudos de Desempenho de IVDVídeoMudanças no IVDR e impactos para fabricantes de IVDServiço de MercadoAcompanhamento Clínico Pós-Mercado (PMCF) da UE para Dispositivos MédicosServiço de MercadoAcompanhamento do Desempenho Pós-Mercado (PMPF) da UE para IVDs


Vamos conversar,
Onde quer que você esteja.
Quer esteja procurando mais informações ou pronto para ser nosso parceiro, estamos aqui para orientá-lo em todas as etapas do processo regulatório.
Fale conosco